


本文是基因慧连续七年发布的《基因行业蓝皮书》(下载链接)连载的第四篇,一览基因技术在肿瘤早筛、伴随诊断和复发监测(MRD检测)的应用,细胞和基因治疗将在下篇单独展开。2025年的《基因行业蓝皮书》启动中(首发中英双语版),欢迎企业/园区/学会合作。
(原文完稿于2024年7月,发布于2024年9月公开的《基因行业蓝皮书(2024-2025)》,略有改动。仅作为研究参考,不作为临床诊疗用)
一、总论
全球正快速进入老龄化社会。根据世界卫生组织(WHO)2022年统计,60 岁以上人口的数量和比例逐年增加(2019年超10亿,2030年预计13亿,2050年预计达到21亿人,占比21.4%)。我国在1999年迈入老龄化社会(参照WHO的标准即60岁以上人口比例超10%,OECD新标准以65岁超7%),2023年迈入中度老龄社会。人口老龄化比例增加最大的是低收入和中等收入国家(2050年将吸纳80%的老年人),这给医疗健康带来重大挑战。
在分子机制层面,年龄的增长累积基因突变从而加速癌症(癌症约等于恶性肿瘤,以下等同称呼)的发生。尽管肿瘤发生呈现低龄化趋势,但癌症主要发生在高龄人群。WHO旗下的国际癌症研究机构(IARC) 2024年预计,约1/5的人在一生中会患上癌症,2022年预估有2000万癌症新发病例和970万死亡病例,癌症5年生存人数约为5350万。在36种常见癌症中,最高发癌症是肺癌、乳腺癌和结直肠癌。根据我国国家癌症中心2024年统计,2022年我国新发癌症病例482.47万,发病率从35-39岁年龄组开始显著增加,在80-84岁达到峰值,平均每年增加1.4%;2022年癌症死亡病例257.42万,全癌种死亡率在40-44岁后显著增加。
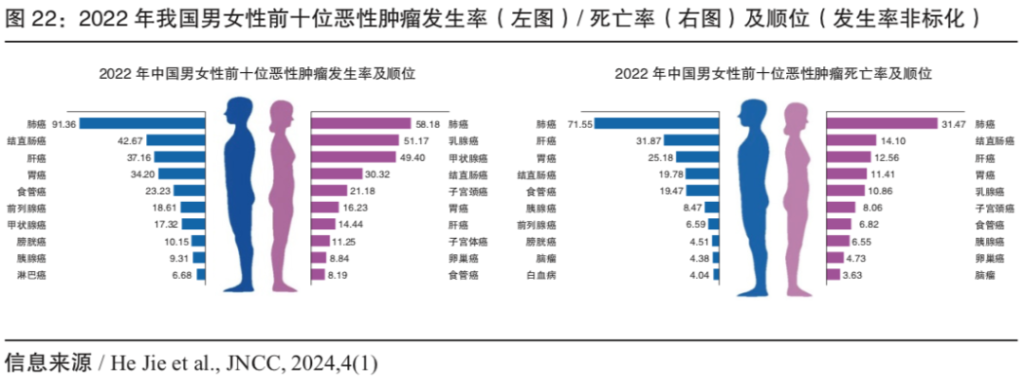
图:2022年我国恶性肿瘤发生率(左图)和死亡率(右图)及顺位(来源/He Jie et al.,JNCC, 2024)
随着癌症防治的战略高度、公众意识和精准医疗技术的提升,我国恶性肿瘤的5年总体生存率从2015年的40.5%上升到2022年的43.7%,但与发达国家仍存在约25个百分点的差距。
癌症的本质是基因异常疾病,由不受控制的的细胞增殖引起。目前癌症治疗以手术(外科切除)、放疗(放射治疗)、化疗(化学药物治疗)为主,还包括免疫治疗等新兴治疗手段。由于癌症细胞具备免疫逃逸、易转移(不具有传染性)等特点,使得病程的预后差。这个问题可以通过分子分型和早筛早诊技术来改善。
由于部分癌种的组织取样困难且在早期难以发现潜在的癌症,使得病理诊断相对受限。随着基因技术逐渐成熟,肿瘤分子分型通过对肿瘤驱动基因变异的检测和特征谱的解析,在临床上可以对肿瘤进行精准的分子分型并挖掘特异关键指标进行预后评价监测。分子分型也推动肿瘤分类由组织形态学向分子特征转变,即一种肿瘤可能有多种亚型并携带不同的突变基因,不同的肿瘤可能携带相同的突变基因,对应在抗肿瘤药物临床试验设计上的篮式试验与伞式试验。同时分子分型也可以发现早期的癌症。
肿瘤与基因多种变异相关,包括:DNA 突变、扩增、RNA 融合、甲基化、TMB(肿瘤突变负荷)、MRD(微小/可测量/分子残留病灶)、MSI(微卫星不稳定性),在不同的病程阶段具有迥异的特征。基因变异可以被免疫组化(IHC)、荧光原位杂交(FISH)、PCR、NGS等技术检测到,为临床诊疗提供分子依据。
总体上,基因检测技术在肿瘤防控方面主要用于肿瘤早筛、肿瘤伴随诊断及预后监测等。肿瘤基因标志物从基因突变发展到TMB、MSI、甲基化等多种类型,从单癌种到泛癌的基因标志物,从肉眼可见的病灶诊断到MRD检测。其中,肿瘤用药及预后监测是临床上较为成熟的应用。目前,美国FDA已批准超过80种针对肿瘤基因突变、覆盖30多种癌症类型的靶向治疗药物和免疫治疗药物,且规定靶向药好免疫药物与伴随诊断试剂同步上市。我国国家药品监督管理局(NMPA)已批准超过18种与肿瘤诊疗相关的基于NGS的体外诊断(IVD)试剂,在LDTs(实验室自建检测,laboratory-developed tests)试点(北京、上海、广州)中也广泛用到临床基因检测。
在基础研究方面,近年各国发起肿瘤基因研究的科学计划和队列研究。2006 年,美国国立卫生研究院(NIH)旗下的国家癌症研究所和国家人类基因组研究所发起了癌症基因组图谱计划(The Cancer Genome Atlas,TCGA),作为国际肿瘤基因组协作组研究计划的最大组成部分,计划绘制一个全面的、多维的、针对多种癌症基因组的图谱,包括基因组、转录组、表观组学测序以及整合分析,并将多组学数据与临床和影像数据相关联;2012 年,TCGA提出泛癌症计划(Pan-Cancer Initiative);2018 年,泛癌症计划发布了 11,000 多例肿瘤患者所患有的 33 种最普遍的癌症类型的泛癌症图谱。2019年,国家人类遗传资源中心组织《中国肿瘤基因图谱计划》(“CGAC”)并启动肺癌、乳腺癌、胃癌等项目;2023年,北京市希思科临床肿瘤学研究基金会(CSCO)发起“中国癌症基因组图谱”项目(“CCGA”)。
在临床应用方面,肿瘤基因检测从单基因到多基因,从单癌种到多癌种到泛癌种,从小panel到大panel、从突变、融合到TMB等多类型标记物,从伴随诊断、早筛到预后的肿瘤全周期管理。在PCR等技术持续成熟应用临床的同时,针对NGS的肿瘤基因检测审批在2017前后加速。NMPA在2019年前后批准十余个靶向NSCLC的小panl,2023年批准首个原研伴随诊断、免疫治疗伴随诊断以及首个NGS 大Panel。
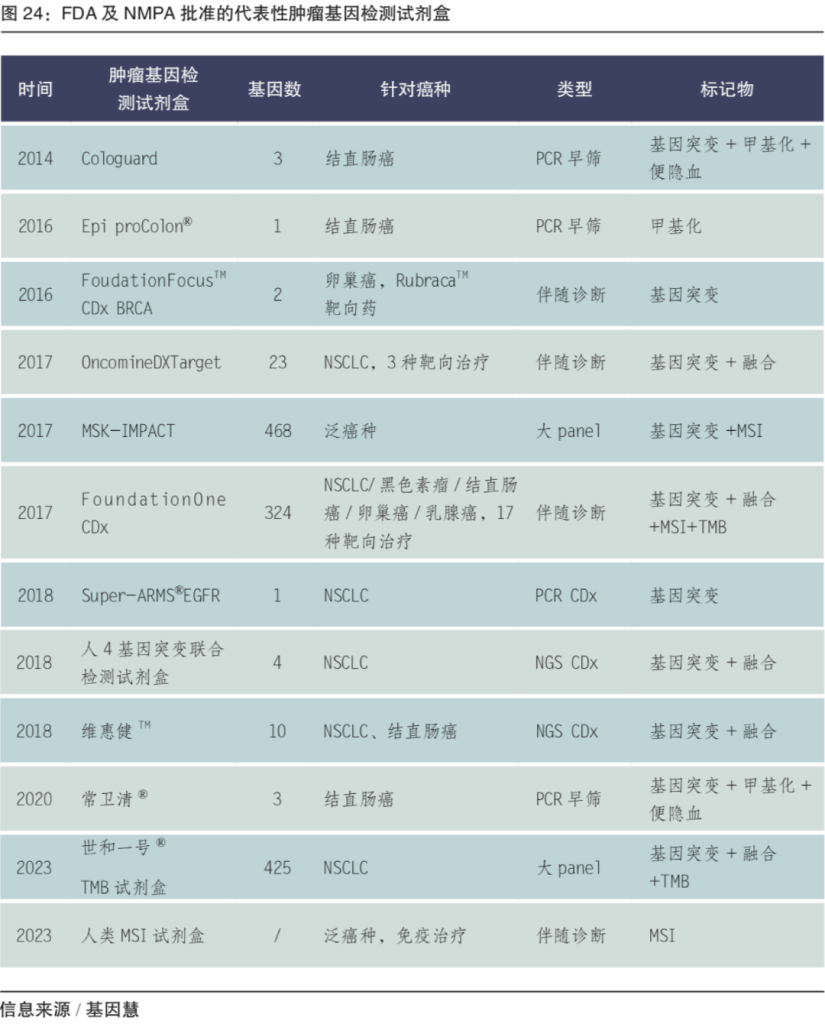
图:FDA及NMPA批准的代表性肿瘤基因检测试剂盒(来源/基因慧整理自公开资料)
在产业方面,由于肿瘤基因检测产品开发周期长且市场尚未完全成熟,肿瘤基因检测企业尚未实现盈利(平均亏损20%及以上);受监管环境以及资本市场低迷等因素的影响,目前国内外肿瘤基因检测企业的发展处于瓶颈期。同时,基于当前的老龄化、靶向药开发以及肿瘤早筛等需求,肿瘤基因检测赛道存在极大未被开发的市场潜力。目前国内上市的针对肺癌的四个驱动基因的靶向药物均被纳入医保。根据NMPA的最新规定,靶向药使用之前均需进行基因检测。中山大学孙逸仙纪念医院2022年在Nature Communications上发布1万例中国患者泛实体肿瘤体细胞突变情况(包含25个癌种和100多个肿瘤亚型),发现64%的中国癌症患者具有临床上可用药的潜在基因突变(PMID: 35871175);2022年CSCO肿瘤生物标志物委员会发起的“中国肿瘤NGS大型应用调研”中,根据30个省市2053个有效样本的调研发现NGS检贯穿肿瘤患者诊疗全程,达到71.3%的比例。
肿瘤基因检测赛道国外代表企业包括Exact Sciences、Guardant Health、Myriad、Grail、Natera等,国内代表企业包括艾德生物、鹍远生物、燃石医学、世和基因、吉因加、海普洛斯、泛生子、华大基因、锐翌生物等。艾德生物作为率先实现盈利的肿瘤分子检测企业,2023年营收10.44亿元(同比增长24%),扣非净利润2.39亿元(同比增长52%)。作为基因检测头部机构的华大基因,2023 年度肿瘤与慢病防控业务营收5.25 亿元(同比增长30%)。作为专注NGS肿瘤基因检测首家上市机构,燃石医学在2023年的重大亏损基础上实现较大跨越,2024年第一季度实现Non-GAAP毛利扣除相关费用后的单季度扭亏为盈。作为海外龙头肿瘤基因企业Exact Sciences在2023年总营收25亿美元(增长20%),其中筛查收入为18.65亿美元,精准肿瘤检测收入为6.29亿美元,官方预计2024年营收约为28.1-28.5亿美元(增长13%)。
根据产业周期,知名的基因产业咨询机构基因慧预计2024-2025年在肿瘤基因赛道可能产生并购或战略投资。此外,国内企业的出海也是肿瘤基因的重点市场。以艾德生物为例,从2019-2023年(除了2021年)在海外市场的增长率均超过35%。
从长周期上,基于药物研发及临床需求的基本面,肿瘤基因检测对监管政策、医保及一级市场投资依赖性较大,由于开发周期长且成本大,目前面临相对较大的经营挑战;我们也看到,产学研专家积极推进共识规范的出台,技术平台的开发机构已深度参与审评标准制定,经济周期叠加产业周期出清了相当比例的同质化低价值的产品及机构;在“AI+BT”的技术及行业跨界融合的刺激下,真实世界数据的积累、大模型及转化研究,正有序地整合生命多组学、患者表型、临床影像及诊疗信息等,未来有望构建特定疾病、特定地区的数据平台以及家庭、个人的数字生命健康中心,为公共卫生药物研发及肿瘤全生命周期管理等提供更精确、更多元的科学依据,逐步展现肿瘤基因检测市场更多的应用场景和更大的市场潜力。
除了科研院校、医疗机构和企业方之外,癌症公益组织在肿瘤防治中发挥着重要的角色,通过肿瘤科普、患者联络、基金会等,推动肿瘤的认知、诊疗及产学研用协作。国内的相关机构包括中国癌症基金会(http://www.cfchina.org.cn)、专注儿童癌症科普的“向日葵儿童”,在线癌症患者教育和院外康复管理平台“觅健”,深耕肿瘤医研企交流及会议培训的“中国抗癌协会肿瘤标志专业委员会”等。在本章节从临床应用角度,基因慧重点阐述肿瘤基因早筛、伴随诊断(用药指导)以及MRD检测(预后监测)等技术、应用及产业(注:基因和细胞治疗将在单独其他章阐述;更多内容欢迎关注及合作基因慧可能将发布的新版《肿瘤基因检测蓝皮书》)。
二、肿瘤(基因)早筛
(一)需求
由于肿瘤细胞的易转移特性,中晚期癌症治疗复杂且致死率高,所以早发现将有效提高患者生存率,改善诊断和治疗服务的可及性和可负担性,提高医疗效率。在宏观层面,肿瘤早筛有助于建立公共卫生基线,降低医疗健康支出,推动医疗工作者对癌症早期的认知和基础研究,加速肿瘤治疗靶点的发现。2021年WHO建议将DNA检测作为预防宫颈癌的首选筛查方法;同时,WHO也提示需注意筛查可能带来的过度诊断和卫生系统成本。
(二)定义
根据WHO的定义,肿瘤的早发现(early detection)分成肿瘤早筛(screening)和肿瘤早诊(early diagnosis)。肿瘤早筛是在健康人群中来识别患有疾病但尚未出现症状的个体,主要筛查部分癌症(或者部分分子特征),需要充分考虑流行病学、覆盖几乎所有目标群体、技术可行性等卫生经济成本及社会效益。对于早筛高风险结果需要临床进一步检查来确诊;而肿瘤早诊侧重于尽早发现有症状的患者,减少晚期才被诊断的患者比例,理论上是针对所有癌症。
肿瘤早筛的手段包括医学影像学(例如 X 光筛查乳腺癌、低剂量螺旋 CT筛查肺癌等)、内窥镜(例如肠镜筛查结直肠癌)、组织活检和细胞学(宫颈刮片筛查宫颈癌等)、肿瘤标志物(蛋白质和酶类、激素、代谢产物、CTC、ctDNA、基因和分子标志物等)。其中,组织活检仍是金标准,ctDNA以及基因等新型标志物越来越多应用到大人群样本队列研究以及临床肿瘤诊疗中。本章节主要阐述基于基因检测的肿瘤早筛(注:如无特别说明,下文提到的肿瘤早筛即基于基因等分子标志物的早筛),核心优势是早发现、覆盖多癌种、用户依从性好以及相对较高的特异性。
对于早筛的疾病范围,WHO的Wilson 和Jungner在1968年提出了十条标准,被NIH等公共卫生健康部门决策沿用至今。
适合筛查的疾病范围标准建议
(Wilson 和Jungner,WHO,1968年)
- 筛查的疾病事关重大的健康问题
- 对确诊的患者应该进行治疗
- 对筛查出的疾病有诊断和治疗的设施
- 筛查出的疾病有一个可识别的潜伏或早期症状阶段
- 提供合适的检测或检查方法
- 检测或检查方法是大众可以接受的
- 充分了解疾病的自然史(不给任何治疗或干预措施)
- 疾病判断的准则应对任何人保持一致
- 筛查、诊断和治疗的费用应在医疗健康的总费用范围内实现经济平衡
- 筛查是一个对方法、中间步骤和结果持续优化的过程
根据WHO的国际癌症研究机构(IARC),衡量大规模癌症早筛项目的关键指标包括参与率、检查覆盖率、进一步评估率与检出率等。

图:大规模癌症早筛项目的关键指标(来源/ IARC,制图/基因慧)
(三)应用
根据美国临床肿瘤学会(ASCO)官网,肿瘤早筛指的针对较高遗传风险和环境风险的人群,进行患病评估和高危人群的识别;建议重点筛查的癌种包括乳腺癌、结直肠癌、宫颈癌、子宫内膜癌、肺癌、前列腺癌等。根据人群患病频率等因素,目前我国启动的癌症早筛项目主要包括女性“两癌”(乳腺癌、宫颈癌)、结直肠癌和肺癌,其次是肝癌、胃癌、食管癌、前列腺癌等。其中,结直肠癌早筛的产业化相对成熟。
从产品模式上,肿瘤早筛的范围从高发单癌、多癌种组合演化到泛癌种。其中,高发单癌的早筛相对成熟,绝大部分多癌种和泛癌种早筛处于临床研究阶段。在临床应用采纳产品时,,需要特别重视早筛产品注册证中所显示的适用范围。目前我国获批的肿瘤早筛产品仅为个位数(且针对高风险人群),临床应用时需与肿瘤辅助诊断产品区分开来。
肿瘤早筛的应用场景主要是医疗机构院内,其次是院外体检中心以及直接面向消费者(C端)的居家检测。不同场景检测核心区别之一是采样方式。采血等侵入式主要在院端和体检端,C端的样本主要是唾液、口腔黏膜、粪便等通过非侵入式采集。目前为响应肿瘤防治,民生工程也是一种重要的应用场景和模式,根据合作机构及检测产品的不同而采取灵活的采样方式,政府作为支付方。
值得关注的是,临床基因检测应用越来越广泛,但未发展到成熟的IVD模式,传统的审评机制带来的滞后效应往往导致产品上市时有被新兴技术迭代的困境。由于目前绝大多数院端等类似机构不具备检测的条件(主要是试剂研发人才、检测团队以及实验室负荷率),肿瘤早筛技术检测基本都在第三方检测机构实施,及市场上实质上采用LDT模式,但LDT尚未有明确的规范。当前,基因检测的样本流、信息流以及支付流未形成闭环,增加相当大的成本及风险。因此,业界对LDT的规范指南及推广有较高的呼声,目前我国有关部门在北京、上海、广州等地的部分医院相对谨慎地有序开展试点;在美国,此前由美国联邦医保及医助服务中心(CMS)对LDT进行专项要求,2024年4月底FDA明确将在未来4年内逐步取消对LDT的一般执法自由裁量权,可以理解将LDT按照类似IVD来管理,除了医疗机构以及其他特殊部门内部开发的LDT可以豁免部分监管,这可能将降低伴随诊断及新药研发的积极性,阻碍中小企业的创新竞争。
(四)技术
在谈到肿瘤基因早筛之前,本报告先介绍肿瘤早筛的各种生物标志物(基因、蛋白、细胞、微生物等(本质是反映肿瘤细胞发生、发展、凋亡过程中的主要特征)。
Douglas Hanahan和Robert Weinberg从2000年起基本每隔10年对癌症主要特征及标志物进行总结,从2011的6个特征增加到2022年14个特征,包括:无限复制潜能(例如端粒酶)、自给自足的生长信号(H-ras基因)、对生长抑制信号不敏感、逃脱细胞凋亡(IGF)、持续的血管生成(VEGF)、组织侵袭和转移的能力、基因组不稳定性和突变(TP53等)、免疫逃逸(PD-1/PD-L1)、肿瘤炎症、能量代谢失调、表型可塑、表观遗传、微生物组、衰老细胞。
基于取样方式,肿瘤基因检测包括以FFPE样本(福尔马林固定石蜡包埋组织,保持细胞完整性)的组织活检以及液体活检两大类。目前组织活检还是金标准,占比超过60%。从肿瘤取样的可及性及依从性,液体活检在肿瘤早筛等领域较为常用。
液体活检的检测对象包括循环肿瘤细胞(CTC)、循环肿瘤 DNA(ctDNA)、外泌体(Exosome)、细胞外 RNA(exRNA)等。其中,ctDNA检测技术相对相对成熟,由于是肿瘤细胞释放到人体循环系统的游离DNA,片段小,含量极低,对技术平台要求较高。从技术平台上,基于PCR的液体活检率先获得临床准入注册证,主要用于单癌种;基于NGS技术的检测主要用于泛癌种,具备更快的发展速度和可能更大的市场前景。ctDNA检测以基因突变标志物为主,按照基因及位点数量层面,依次可以选择小Panel、大Panel、WES和WGS等;除了基因突变,甲基化检测近年发展较快(国内有10余个产品获批);片段化特征检测是近年的新兴技术。
根据中华医学会检验医学分会、国家卫生健康委员会临床检验中心共同制定的《液体活检在临床肿瘤诊疗应用和医学检验实践中的专家共识》,对于检测患者ctDNA是否含有已知的、单个靶向治疗敏感或耐药型突变时建议使用ARMS(扩增阻滞突变系统PCR);检测已知的部分或全部临床药物治疗靶点谱或耐药指示靶点谱,或发现患者基因未知突变、探索临床价值与相关机制时建议使用NGS。
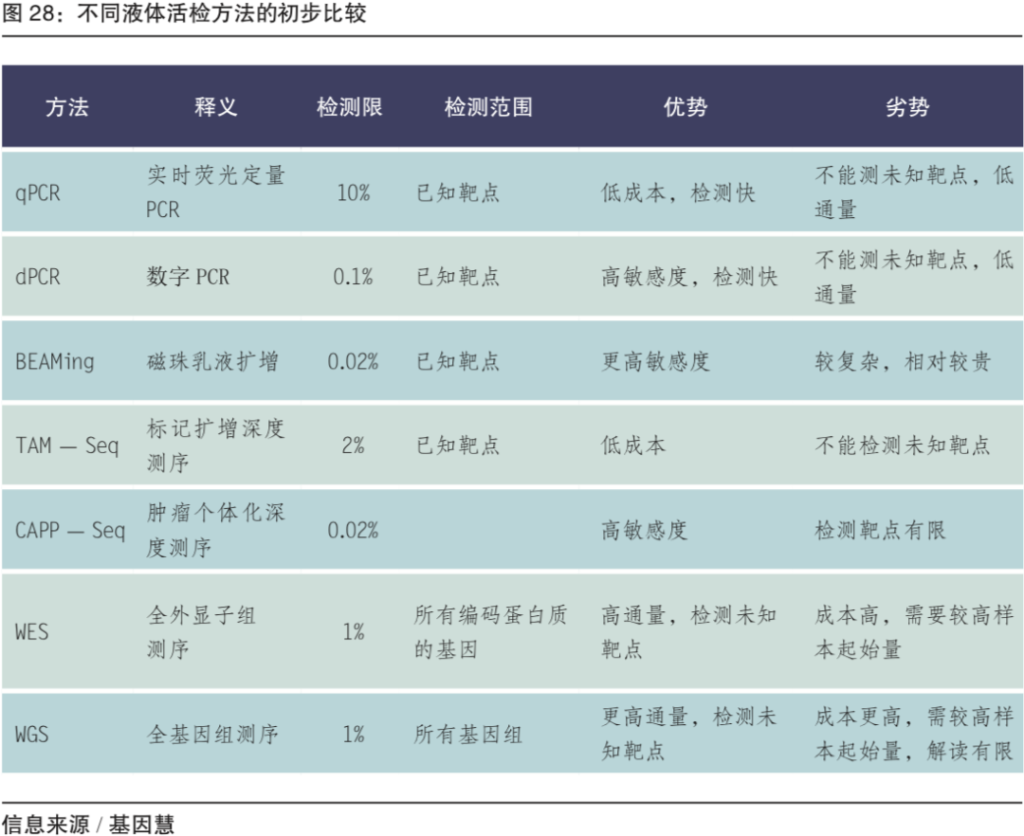
图:不同液体活检方法的初步比较(来源/基因慧整理自公开资料)
其他液体活检方法也在快速发展中,2021年NMPA批准国内首张CTC三类注册证设备“CytoSorter®循环肿瘤细胞检测系统”(基于纳米微流控和免疫捕获),目前尚无对应早筛试剂盒获批;外泌体是一种由活细胞分泌到细胞外的小囊泡(直径约为30-150nm),由于其组织特异性以及活细胞动态信息反应的敏感性,理论上能提高阳性预测值(PPV)。2024年1月由思路迪诊断研发的外泌体卵巢癌检测试剂盒获国家药品监督管理局批准上市。根据思路迪官方公布的四个临床中心开展的注册临床研究数据显示,在上皮性卵巢癌和附件良性包块的鉴别诊断中特异性达到90.2%,在I期卵巢癌中诊断的敏感性89.7%。
根据国家药品监督管理局医疗器械技术审评中心2021 年发布的《结直肠癌筛查产品国内外现状及临床评价要求》,疾病筛查类产品的临床试验设计应针对预期的筛查人群进行前瞻性入组,要评价考核检测试剂的临床灵敏度、特异度、PPV(阳性预测值)和 NPV(阴性预测值) 等指 标,特别是灵敏度和 NPV 必须达到较高的水平才能作为筛查产品上市。这对于早筛产品研发提出较大挑战,因为早期肿瘤患者 ctDNA丰度较低,需要进一步降低最低检测限和提高检测灵敏度;同时,前瞻性研究要求开展大样本队列,对投入成本有一定要求(数千万元至数亿元)。以业内标杆产品之一的Cologuard为例,根据Exact Sciences公司在2024年J.P.摩根健康大会上的报告,2025将发布的下一代产品的结直肠癌检测的特异性为91%,对癌症和成熟癌前检测敏感性分别是94%和43%。
研究者为解决ctDNA丰度较低等问题,发明了多种标志物联合检测方式,包括点突变、甲基化、片段化特征、拷贝数变异等多种基因变异特征以及DNA、RNA和蛋白的联合检测。为评估多癌种的不同早筛技术,2024年2月,美国国立卫生研究院启动一项临床试验CSRN,组织8个小组在7家癌症机构招募24,000通过随机对照试验来分析有前景的癌症筛查新兴技术的利与弊,并确定如何更好地应用临床。
多种标志物联合检测较为成熟的范例是“基因点突变 + 甲基化 + 蛋白”联合检测。2014 年 FDA 批准全球第一个结直肠癌基因筛查联合粪便隐血检测产品 Cologuard,采用的技术路线是“NDRG4 和 BMP3 基因的甲基化 + KRAS 基因的点突变 + 大便隐血中的血红蛋白检测”,实现结直肠癌筛查 92.3% 的灵敏度。2020 年我国NMPA批准的首个肿瘤早筛产品“常卫清”采用类似的技术路径,即“体外粪便样本中的 KRAS 基因突变 +BMP3 和 NDRG4 基因甲基化 + 血红蛋白检测”, 根据诺辉健康公布的资料,根据Clear-C的I期临床试验结果,常卫清对于肠癌的检测灵敏度为95.5%;对于进展期腺瘤的检测灵敏度为63.5%;对结直肠癌的阴性预测值高达99.6%。除了多靶点多维度检测之外,超深度靶向测序 ctDNA 突变也是解决低丰度富集的方法,但增加对应的成本。
除了以上技术,组织特异性的甲基化图谱和循环蛋白标志物等方法,在肿瘤组织溯源方面也发挥着重要作用。
(四)产业
从市场需求及技术可及层面,肿瘤早筛是未来基因产业中规模最大的赛道之一。同时,肿瘤早筛具备较高的研发门槛(包括技术门槛及大规模队列研究的成本)。2020年,肿瘤早筛赛道在产品未成熟前被过热炒作,在获得较高期望值后回归理性,目前处于调整期。
在渠道方面,肿瘤早筛的核心对象针对健康人群中的高风险人群(未发现明显的疾病表型),主要商业渠道是体检机构以及高端医疗健康中心(B2C),C端居家检测市场尚不完全成熟;民生工程是国内的集中检测的主要渠道之一,短期内对区域头部机构培育和肿瘤早筛普及有较大推动作用,但不适宜长期的商业战略(公共卫生支出有限,且易形成路径依赖)。LDT的准入是技术创新及商业发展的重要平衡方式,作为企业和医院、药企等合作实施的可选项,目前在试点中。
在支付方面,肿瘤早筛产品目前处于商业化早期,还没有纳入医保。国外主要以商业保险覆盖,国内主要是受检者自费。
在产品战略方面,包括横向产品组合和纵向全周期管理。横向战略,即从产品组合切入,以PCR为主,结合高通量测序、单细胞、CTC、外泌体检测等多种技术进行肿瘤早筛,多元技术扩大应用场景及用户规模;纵向战略,即从肿瘤全周期管理切入,以NGS技术为主,贯穿早筛、伴随诊断、MRD等应用,聚焦临床大客户以及药物研发服务两大路线。
国外肿瘤早筛企业包括Exact Sciences,Grail,Freenome,Guardant Health等,国内肿瘤早筛企业包括诺辉健康、鹍远生物、燃石医学、泛生子、锐翌生物、吉因加、桐树基因、华大基因、和瑞基因、康立明、艾米森、博尔诚、达健医学、基准医疗、为真生物、奕谱生物等。
以标杆企业Exact Sciences为例,2014 年推出肿瘤早筛明星产品Cologuard,售价约500 美元(对比肠镜检查平均费用约 2125 美元),销售数量从 2014 年 4000 份开始至 2022 年累计 1400 万份(2022年400 万份)。2023年筛查营收18.65亿美元(同比增长31%),公司预计2024年筛查业务营收为21.55-21.75亿美元(增长率调低为16%)。2023年Exact Sciences的净利润为负数,调整后税息折旧及摊销前利润(Adjusted-EBITDA)为2.19亿美元。
另一个标杆企业是Grail。Illumina孵化的肿瘤早筛企业Grail于2024年正式分拆。Grail自2016年成立至今,运营总费用预计超过25亿美元(公司公布预计2026年产品上市估计还需10亿美元),临床试验总人数累计38.5万人,2023年总费用7.65亿美元。从2022年Q4到2024年Q1,每季度平均营收约2300万美元。在2024年美国癌症研究协会(AACR)年会中,Grail公布的Galleri临床试验结果显示,产品特异性为99.5%,敏感性为51.5%,假阳性率为0.5%,阳性预测值(PPV)为44.4%,组织溯源准确率为88.7%。
国内肿瘤早筛赛道的商业化在 2020-2021 年达到阶段性高峰。肿瘤早筛头部企业之一鹍远生物于2020年获10亿元B轮融资,刷新早筛赛道融资记录,2024年获得扬州市区各级国资战略投资的过亿元融资;2021 年,国内早筛首证“常卫清”的获批给市场注入极大动力,类似获批产品包括锐翌生物的常易舒(检测 SFRP2 和 SDC2 基因甲基化)、艾米森的艾长康(检测 SDC2 和 TFPI2 基因甲基化)以及康立明生物的长安心(检测 SDC2 基因甲基化)等,在各个区域市场获得较大增长。
2023-2024年,肿瘤早筛产业进入调整期。燃石医学的2023年财报不及预期(2023年净亏损6.54亿元),泛生子完成私有化退市(交易股权价值约1.26亿美元,2022年6.51亿元,归母净亏损8.08亿元),世和基因撤回科创板上市申请,同时我们可以看到,产品方面,国内肿瘤大Panel试剂盒获批,FDA明确了多癌种早筛即将准入,这对未来更大规模的临床应用市场释放利好信号;产业运营方面,世和基因2023年实现盈利(据公开信息),燃石医学在2024年第一季度实现了Non-GAAP毛利扣除相关费用后的单季度扭亏为盈;Freeome于2024年2月获得罗氏领投的2.54亿美元。技术迭代及头部企业市场拓展等展示出肿瘤早筛赛道的基本价值面和较大未满足需求,待政策刺激及资本回暖后将再次进入高速发展的周期。
在肿瘤基因早筛领域,基因慧建议未来关注以下方面。
技术层面:启动多中心临床研究,开展结合多种标志物、多组学、泛癌种的早筛早诊队列研究。例如2024年初,北京大学医学部牵头,联合附属10家三甲医院,预计用6年时间,通过对数万例入组人群的血浆游离DNA、RNA和蛋白质大规模鉴定,发现具备高诊断性能的癌症诊断标志物,覆盖癌种包括肺癌、结直肠癌、胃癌等22个中国主要高发癌症。
模式层面:建议关注收入/销售费用比、出海、民生工程及并购。以Exact Sciences为例,2019 年以 28 亿美元收购早期浸润性乳腺癌的基因检测公司 Genomic Health,同年使得旗下的Oncotype DX 乳腺癌复发评分产品被德国纳入医保,2022 年Exact Science 将 Oncotype DX 系列产品之一GPS test(基于基因检测的前列腺风险评分)以 1 亿美元出售给 MDxHealth 公司;2020 年,Exact 以分别以21.5 亿美元(以及股票兑价)和4.1亿美元收购液体活检企业Thrive Earlier Detection和甲基化检测企业 Base Genomics。在运营优化方面,以Exact Science为参考,2022年的收入/销售费用比为2.46,国内企业仍有一定差距。诺辉的DTC/B(保险公司、健康管理公司、电商平台)和民营体检渠道模式,充分利用投资机构资源,以结算价让利换量。锐翌生物与海得威医药、轻松集团合作多元化渠道推进肠癌早检。
制度层面:建立产业园及区域中心支持企业组团“出海“,通过国资投资、地方政府参与等形式支持大规模前瞻性队列研究。目前,莱蒙健康、泛生子、基准医疗、燃石医学、奕谱生物、鹍远生物、中精普康等企业的肿瘤筛查/诊断产品获FDA突破性医疗器械。2023 年,鹍远生物与马来西亚医疗器械公司 Vanguard MedTech 达成肠癌多基因甲基化检测技术 ColonAiQ®的战略合作,联合美国突破基因组学(Breakthrough Genomics)公司在加利福尼亚州启动胰腺癌液体活检技术 PDACatchTM癌症早期筛查和检测服务;目前数个肿瘤早筛大规模前瞻性队列在进行中,为肿瘤早筛赛道积累雄厚的样本、数据及研究基础,未来需加强大样本队列的成果转化机制和数据合规共享。例如,2021年鹍远生物与扬州市邗江区合作启动全区12万名40-74岁的户籍居民的结直肠癌筛查民生工程,2022年世和基因与南京市江北新区公共卫生服务中心合作启动约10 万例金陵队列,2024年华大基因与哈尔滨市合作80万适检人群消化系统肿瘤及“四高”检测等。
三、肿瘤伴随诊断
(一)需求
靶向治疗是肿瘤治疗中除了手术、放疗、化疗之外的重要方法。为了给临床癌症患者筛选安全有效的靶向治疗方案,以及在药物研发中筛选对特定方案有效的患者,即诞生了伴随诊断,基于生物标记物进行个性化诊疗。2015-2019年间,欧盟和美国批准的药物平均有65%基于生物标志物进行研发,伴随诊断产品已在我国在内的多个国家地区纳入医保。包括FDA以及NMPA等机构均发布了与抗肿瘤药物同步研发的伴随诊断试剂的相关规定。
(二)定义
伴随诊断(Companion Diagnostics, CDx)从英文直译而来,字面意思是伴随着治疗方案选择进行的诊断; 从作用上近似于用药指导,从基因等标志物等信息来确定患者是否适合某种特定的药物或治疗方案、可能的副作用以及监测治疗反应。即在合适的时间为合适的患者在合适的剂量下使用合适的药物,为药物安全性及有效性提供IVD信息。
伴随诊断和药物基因组类似,但本质不同。伴随诊断是一种选择治疗方案的检测手段,相关检测试剂盒产品是医疗器械。药物基因组研究范围更广,研究个体基因差异如何影响药物的作用,包括药物的有效性、药物副作用、药物代谢(与计量调整有关)等。
目前,临床成熟应用的伴随诊断标志物包括DNA突变、基因融合、PD-1/PD-L1;初步应用的甲基化、MSI(微卫星不稳定性)、TMB(肿瘤突变负荷)、MRD(微小残留病灶)、HRD(同源重组缺陷)等。
(三)应用
全球首个伴随诊断产品在1998年获批。2011-2013年,美国、日本及欧盟分别发布伴随诊断相关指南,我国于2020年发布首个伴随诊断的指导原则。
伴随诊断产品临床准入的里程碑
- 1998 年,FDA 批准首个伴随诊断试剂:Dako公司(Roche)的 Herceptest;
- 2011 年,FDA发布《体外伴随诊断试剂指导原则(草案)》》并于2014年正式发布;FDA批准首个基于 PCR 的 CDx:罗氏诊断的 cobas 4800 BRAF V600 突变检测;
- 2013年,日本药品和医疗器械管理局(PMDA) 发布《伴随诊断试剂与药物申请审批通知》。截至2024年5月17日批准了针对40种疾病及病症的CDx产品,产品来自Roche、Myriad、FMI、Guardant Health等机构;
- 2016 年,FDA 批准首个基于 NGS 的伴随诊断产品:FMI的 FoundationFocus CDxBRCA ;
- 2017年,欧洲药品管理局(EMA)在新版体外诊断试剂法规(2017/746/EU)首次明确CDx的定义(之前以CE认证流通),在2022年正式实施后(2022-2024陆续更新)批准首个CDx;
- 2017 年,FDA 批准首个泛癌种(覆盖所有实体瘤)大 Panel 伴随诊断试剂:FoundationOne CDx (F1CDx) (324 个基因的突变、融合、MSI、TMB);
- 2018 年 1 月,我国首个伴随诊断产品获批:艾德生物的 Super-ARMS®EGFR(基于 PCR);
- 2018 年 7 月,我国首个基于NGS技术的肿瘤伴随诊断产品获批:燃石医学的人 EGFR/ALK/ BRAF/KRAS 基因突变联合检测(可逆末端终止测序法);
- 2018 年 11 月,我国首个 NGS 泛肿瘤伴随诊断获批:艾德生物的人类10基因突变联合检测试剂盒(可逆末端终止测序法);
- 2019 年 6 月,北京市将伴随诊断项目纳入乙类医保;
- 2019 年 8 月,我国首个 PD-1 单抗伴随诊断获批:罗氏诊断的 PD-L1 IHC 22C3 pharmDx;
- 2020年 8月,FDA批准首个基于NGS技术的液体活检伴随诊断产品:Guardant 360 CDx;
- 2020年 8月,我国国家药监局医疗器械技术审评中心(CDME)发布首个伴随诊断的指导原则《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》;
- 2021 年 11 月,我国CMDE发布《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》和《使用体外诊断试剂境外临床试验数据的注册审查指导原则》;
- 2022 年 3 月,我国批准首个国产 PD-L1 检测试剂盒(免疫治疗药物伴随诊断): 艾德生物的“PD-L1抗体试剂(免疫组织化学法)”;
- 2022 年 6 月,我国CMDE 发布《与抗肿瘤药物同步研发的原研伴随诊断试剂临床试验注册审查指导原则》。
根据知名基因产业咨询机构基因慧统计,截至2024年5月1日FDA共批准61个伴随诊断产品,对应71种药物(及组合)和31种癌种(同类合并),前五大癌种是非小细胞肺癌、乳腺癌、结直肠癌、黑色素瘤和急性髓细胞白血病等;前五大生物标记物是EGFR、HER2、BRAF、KRAS 和PD-L1 。
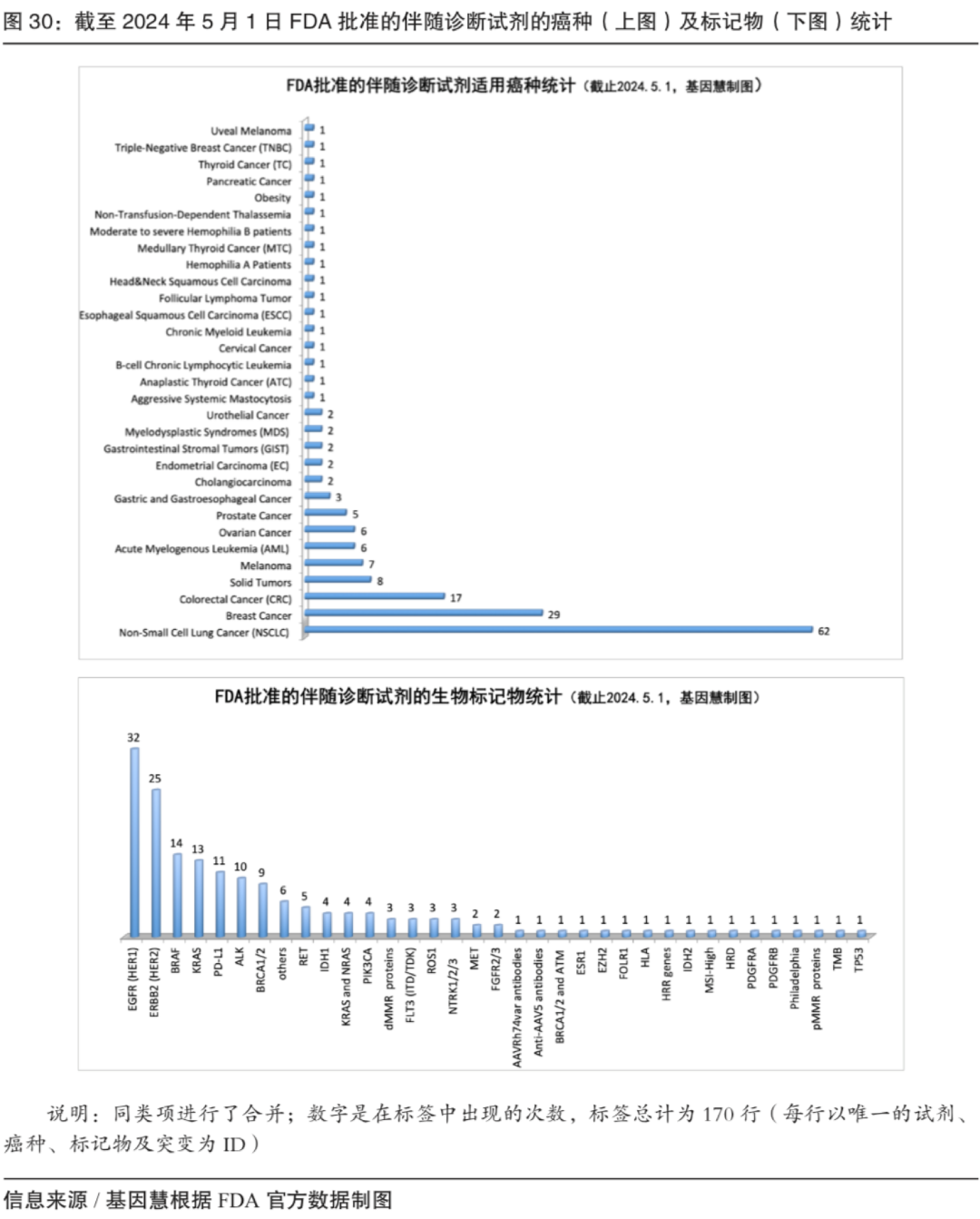
图:截至2024年5月1日FDA批准的伴随诊断试剂的癌种(上图)及标记物(下图)统计(来源/基因慧根据公开数据制图)
伴随诊断的开发方法包括同步(Co-development)和桥接(Bridging)以及跟随(follow-on)。伴随诊断企业与药企通过同步研发实现更经济的同时获批,这也是目前监管部门鼓励的,但费用以及样本要求高;如果没有注册进度完全匹配的伴随诊断研发,通常是采用CTA(Clinical Trial Assay)筛选入组,通过桥接方式开发,来评估药物在CDx定义的人群中的药效,复杂度更高,样本量要求大。更细分的情况,可以分为以下四种开发策略。
在美国、欧盟及澳大利亚等国家,通常把伴随诊断(CDx)归类为III类IVD,CDx产品与相关药物同时开发注册,比在药物批准后注册的时间更短。日本把CDx归类为II类或III类医疗器械,注册时间大约是9个月,基本都可以报销。
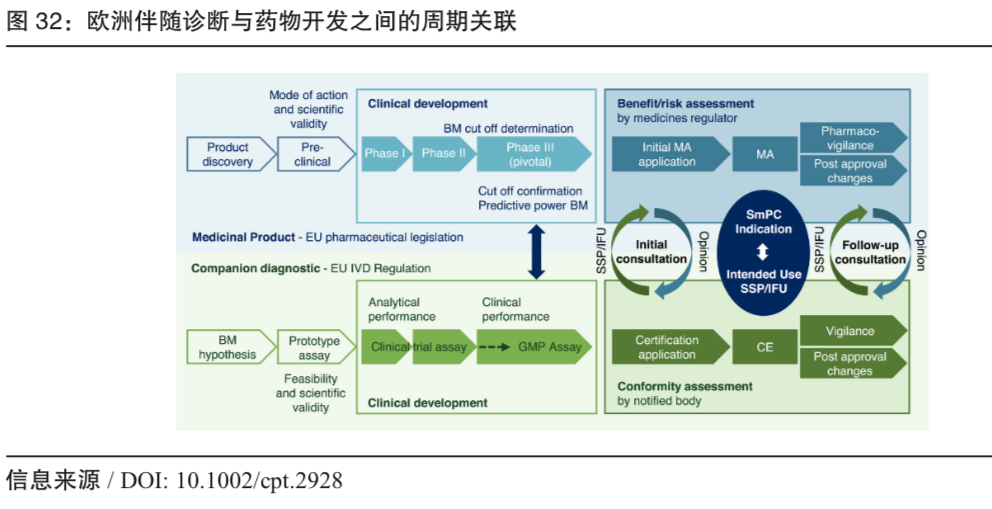
图:欧洲伴随诊断与药物开发之间的周期关联(来源/ DOI: 10.1002/cpt.2928)
(四)技术
不同技术各有优劣,通常根据生物标志物的类型、成本、周期等选择不同的伴随诊断技术平台。针对蛋白质生物大分子的伴随诊断技术平台主要是免疫组化(Immunohistochemistry,IHC)、流式细胞、化学发光等;针对基因等小分子的伴随诊断平台包括荧光原位杂交(Fluorescence In Situ Hybridization,FISH)、PCR和NGS等,此外,Sanger测序、单分子测序、单细胞测序等新兴技术也有相应的应用。本章节以针对基因标记物为主进行分析。
在获批的伴随诊断产品中,较常用的技术平台是PCR,FDA直到2020年才批准首个NGS液体活检伴随诊断产品,但NGS以高通量和新基因发现等特点而以逐年增加比例被采用。根据国家药品监督管理局医疗器械技术审评中心,基于NGS检测人FFPE组织样本的肿瘤基因变异检测伴随诊断试剂,检测的变异基因及位点主要包括两类:按照相关指导原则具有伴随诊断临床证据的为一级位点;尚无明确伴随诊断意义,但国内外权威指南明确地中提出具有临床意义且临床医生可应用的为二级位点。与抗肿瘤药物同步开发的且具有临床试验证据的为二级位点,药物上市后变更为一级位点。针对血浆等游离DNA检测的伴随诊断试剂无明确规定。
(四)产业
肿瘤伴随诊断以“LDT+IVD”联合的形式开展业务合作,主要渠道是医院及药企。与肿瘤早筛侧重于 LDT 模式不同的是,由于伴随诊断的药物疗效评估用途以及在研发及注册上较为成熟,目前在院端侧重IVD模式,和药企合作的主要方向也是基于IVD的研发及注册路径。在产品未取得注册制证前采用LDT模式,当下这种情况居多。
基于商业模式,伴随诊断的支付方主要是院端患者及药企,和医保有较大关联。目前我国主要仅以北京为代表的城市将肿瘤基因检测纳入医保(基于PCR技术)。同时我们看到,国家卫健委颁布的《新型抗肿瘤药物临床应用指导原则(2018年版)》明确指出:对于有明确靶点的药物,须遵循基因检测后方可使用的原则。以肺癌为例,目前国内上市的针对肺癌的驱动基因 EGFR、ALK、ROS1、BRAF 的靶向药物均被纳入医保报销范围之内。这对于伴随诊断的开发和纳入医保具有积极的促进作用。
在海外,日本对Myriad BRACAnalysis和FoundationOne等产品纳入报销,其中Myriad BRACAnalysis可报销202,000日元。我国企业艾德生物的肺癌ROS1基因检测伴随诊断产品自2017年纳入日本医保。美国于2017年批准FMI的F1CDx的同时纳入医保,CMS明确类似IVD纳入医保的产品适用于患有晚期癌症(即复发,转移或晚期IV期癌症)且寻求进一步治疗的受益人。在欧洲,除了比利时、澳大利亚有明确的报销政策(CDx与药物同时报销),大多数欧洲国家的CDx医保政策仍在推进中。
伴随诊断领域代表(以产品获证为主)的国外企业包括FMI(Roche),Guardant Health,Myriad、Dako、Qiagen、Abbott、Ventana等,国内代表企业包括NGS领域的燃石医学、吉因加、泛生子、世和基因、桐树基因、海普洛斯、元码基因等;PCR领域的艾德生物、为真生物、透景生命、飞朔生物等。在业绩表现上,以罗氏诊断为例,2023年在病理诊断领域营收增长14%,比整体营收增长率多8个百分点,增长主要来自伴随和高级染色业务的增长。燃石医学的2024年第一季度的财报显示,在药企合作业务上的营收2060万元(同比下降29%),新合同签约额达到2.18亿元(同比增长190%),反映了肿瘤伴随诊断领域的较大增长潜力。
在肿瘤伴随诊断领域,未来建议关注以下方面。
技术层面:需要进一步规范各项技术的证据优先程度、生物标记物的阈值,来提高伴随诊断产品在临床上的通用性。建议关注的新兴伴随诊断技术:数字PCR、冰冻免疫组化、多重荧光免疫组化、质谱流式、单细胞测序等。
模式层面:建议临床机构加大重视生物标志物等新兴技术的推广,相关机构加大医研企的对话和融合等。根据2020年一项针对新诊断的晚期NSCLC患者的大样本研究发现,超过1/5患者在一线治疗前未接受指南推荐的靶点基因检测,而现成的靶向药治疗方案可以覆盖超过3/5的晚期NSCLC患者。
制度层面:建议进一步出台促进靶向药的研发的法规,增加肿瘤患者更多可治疗方案的选项;鼓励企业联合医院或牵头LDT,为临床医生提供提供更多元、精准的分子检测信息。
四、MRD检测
MRD有时也被归为伴随诊断,但由于在复发监测的独特价值成为近年热门,故单独阐述。
(一)需求
MRD是导致肿瘤复发的主要原因之一,可以通过ctDNA等分子标记物进行早期监测,是血液系统恶性肿瘤临床常用检测手段,近年延伸到实体瘤应用的探索。
2017年,斯坦福大学Maximilian Diehn在Cancer Discovery上发布的研究成果显示,在94%的可评估的肺癌复发患者中,基于ctDNA的MRD在第1次治疗后的血样中就被检测出来,通过MRD检测发现72%患者,比影像学检查平均提前约 5.2 个月(PMID: 28899864);2022年华西医院联合发起的肺癌预后前瞻队列研究LUNGCA显示,术后关键点MRD检测对RFS预测的相对贡献度高于TNM分期等临床变量的总和,长期监测MRD相较于传统影像学提前273天发现复发,治疗后监测期ctDNA MRD阳性预测值为100%,阴性预测值为91.8%。
(二)定义
MRD 指微小残留病灶(Minimal Residual Disease),也被称之为分子残留病灶(Molecular Residual Disease)或可测量残留病灶(Measurable Residual Disease),是指经过治疗后,影像学或传统实验室方法不能发现,但通过液体活检发现的肿瘤来源分子异常,代表着肿瘤的持续存在和临床进展可能。
早在 2007 年,英国贝尔法斯特女王大学的 Alexandra Irvine 和 Mary Mcmullin 对此做过描述(PMID: 17288299),在慢性髓系白血病患者接受治疗后,一小部分会复发,复发源于持续存在的、含量极低、仅能通过分子检测的恶性肿瘤细胞,即 MRD。国内外的MRD队列研究可以关注DYNAMIC、GALAXY、TRACERx、MERMADI、CATHAYA、MEDAL等队列。
(三)应用
MRD作为围术期治疗效果和复发监测的重要方法,对早中期的癌症患者预后监测非常重要,也为个性化治疗方案和优化随访管理提供科学依据。
早在2018年,MRD的血液瘤NGS产品已获得FDA批准,灵敏度达到10-6,即可以检测到低于1/100万个浓度残留癌细胞。MRD在实体瘤方面应用还在不断突破,目前FDA授予部分产品突破性设备认定,国内多个实体瘤MRD在临床试验中,包括结直肠癌、非小细胞肺癌、乳腺癌、胃癌等,并发布多个MRD专家共识。
MRD应用发展的里程碑
- 2018年,FDA批准Adaptive Biotechnologies公司ClonoSEQ®,基于多重PCR扩增BCR/TCR VDJ克隆重排的NGS方法用于对急性淋巴细胞白血病(ALL)或多发性骨髓瘤(MM)患者进行MRD检测;
- 2019年,FDA授予Nater公司的产品Signatera™突破性设备认定(BDD),用于定量检测癌症患者术后血浆ctDNA;2021年再次授予Signatera两个BDD,作为两个癌症药物III期临床试验的伴随诊断;同年,美国Inivata公司的RaDaRTM 实体瘤MRD检测产品获得FDA BDD认定;
- 2020年,FDA再次批准ClonoSEQ ®,用于慢性淋巴细胞白血病(CLL)患者的MRD检测;
- 2021年3月,我国首个MRD共识《非小细胞肺癌分子残留病灶专家共识》在第18届中国肺癌高峰论坛上发布;
- 2021年11月,国家药监局药审中心发布《多发性骨髓瘤药物临床试验中应用微小残留病的技术指导原则》;
- 2024年4月,FDA肿瘤药物咨询委员会(ODAC)根据两项独立的研究EVIDENCE和I2TEAMM的结果,宣布同意将微小残留病灶(MRD)作为多发性骨髓瘤药物研发的临床试验加速批准终点;
- 2024年11月,中国医学科学院北京协和医院吴焕文教授牵头发布《实体瘤分子残留病灶(MRD)检测共识》.
(四)技术
CTC(循环肿瘤细胞)和ctDNA(循环肿瘤DNA)是MRD检测的主要生物标志物。其中ctDNA近年成为焦点,主要检测基因突变(SNV、InDel、CNV、SV)、表观遗传学改变(甲基化)等,需要关注和肿瘤体积大小相关的ctDNA突变频率(VAF)以及不同癌种检测的MRD阈值。
MRD ctDNA检测的技术包括实时PCR(qPCR)、数字PCR(dPCR)和高通量测序(NGS)。流式细胞术(Flow Cytometry, FCM)作为重要的技术手段可以直接检测肿瘤细胞的MRD,灵敏度可以达到10-3~10-4,是当前临床血液瘤MRD检测常用的方法;针对实体瘤,在ctDNA丰度≥0.02%的条件下,可以采用液体活检的方法进行MRD检测,目前主要采用NGS进行实体瘤MRD ctDNA检测。由于肿瘤细胞的高度异质性及含量较低的突变,因此测序深度较高(共识建议至少30,000x甚至100,000x),要注意进行背景噪音纯化。
MRD检测的技术路线包括Tumor-informed assays和Tumor-naïve assays。前者主要是参考组织突变信息进行定制化panel的超深度测序,后者是固定panel或WES测序。
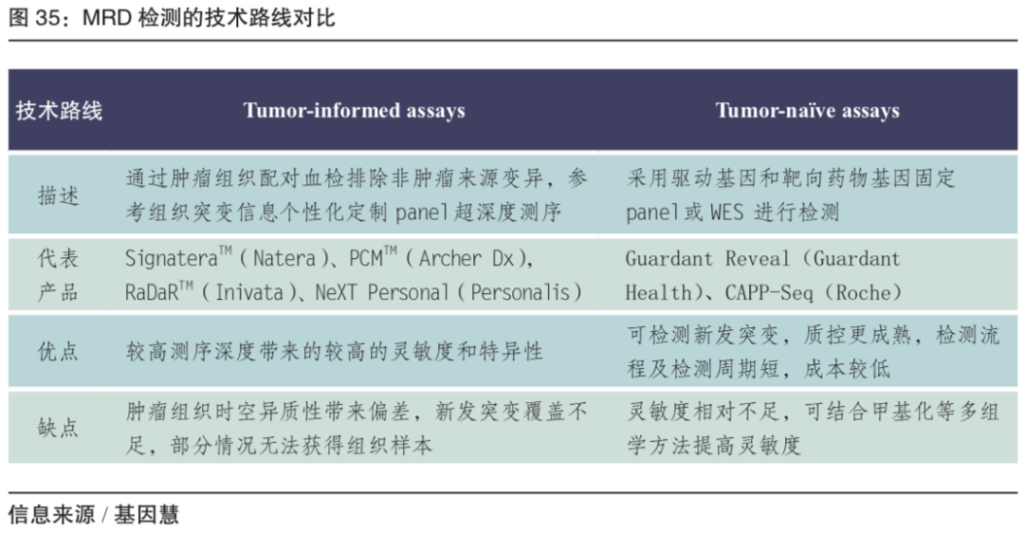
图:MRD检测的技术路线对比(来源/基因慧)
需要关注的是,2024年ASCO会议上发布的临床试验COBRA结果,在针对“临床低危&ctDNA+”的预后研究不及预期;在2024年v1版的结直肠癌NCCN指南中提及不推荐在临床试验之外使用ctDNA进行预测。业界认为这与技术路线可能有关,在技术持续升级的过程中对MRD的价值仍保持较高信心。例如,2024年,吉因加完成与吴一龙教授合作关于非小细胞肺癌早中期、局晚期和晚期MRD研究,分别在Cancer Discovery、Cancer Cell、JAMA Oncology上发布,发现晚期驱动基因阳性肺癌的患者,原本连续用药模式变为密切监测下间断用药模式,总体患者PFS(无进展生存期)达18.4个月。
除了NGS平台,基于PCR的多基因甲基化技术近年也展现出临床应用潜在价值。鹍远生物联合复旦大学附属肿瘤医院、上海交通大学医学院附属仁济医院开展首个基于PCR的血液ctDNA多基因甲基化技术应用结直肠复发监测,发现术前ctDNA阳性患者,术后复发概率高于术前ctDNA阴性患者(22.0%>4.7%);根治性切除术后一个月,ctDNA阳性患者复发概率是阴性患者的17.5倍。值得关注的是,基于PCR的ctDNA多基因甲基化检测,在检测费用和报告周期上有较大提升。
(五)产业
目前MRD检测的付费主要以用户个人支付为主,在部分地区被纳入医保。Adaptive Biotechnologies的clonoSEQ在获批后的第二年(2019年)被纳入美国医保。Natera公司的Signatera产品的膀胱癌复发监测、乳腺癌复发监测分别于2022年、2023年获得美国医保覆盖。Signatera和Guardant Health公司的Guardant Reveal产品在2023年先后获得美国商业保险蓝盾公司(Blue Shield of Louisiana)的承保。
MRD领域代表性的国外企业包括Naera、Guardant Health、Freeome、Adaptive Biotechnologie、NeoGenomics等,国内企业包括鹍远生物、世和基因、吉因加、臻和科技、华大基因、至本医疗等。
MRD赛道的业绩在2023年整体表现突出。根据公开信息,Guardant Health 表示2023年MRD检测量同比增长90%,预计全美1500万名患者和200亿美元的市场;血液癌症筛查作为未来的两大业务重点之一。Natera预计2024年全年营收14.2-14.5亿美元,2024年第一季度营收同比增长52%,MRD产品Signatera获得空前的增长,单价提高到1000美元上。专注MRD的Freeome公司在2023年完成由罗氏诊断领投、Quest等跟投的2.54亿美元的融资,强调了基于多组学、机器学习和真实世界数据的多组学平台上推进癌症早期检测。
2020年,我国首个肺癌商业化MRD检测上市——臻和科技的“朗微博”。2021年,华大基因独家将Natera的 Signatera产品引入中国,通过国产测序平台10万x超深度测序,检测限达0.009%,2023年肿瘤复发监测业务营业收入同比增长约 58%。
在MRD检测领域,未来建议关注以下方面。
技术层面:进一步迭代ctDNA、甲基化等多元技术的产品研发,加大前瞻性队列研究投入以进行技术验证和临床应用验证。
模式层面:MRD与其他技术组合构建肿瘤基因检测综合方案。2020年, Exact Sciences收购Ashion(希望之城癌症治疗中心转化基因组研究所转化)获得MRD检测技术TARDIS。2021年,NeoGenomics以3.9亿美元收购Inivata切入MRD检测。
制度层面:加速MRD的临床准入,基因慧预计至少需要2年时间。具备核心MRD技术的产品及企业标的是极具价值投资并购的标的。
【致谢】
黄荷凤院士、卢光琇教授作序推荐。感谢星云基因、华大智造、赛福基因、鹍远生物对《基因行业蓝皮书(2024-2025)》的大力支持,得以公开发布和传播。
【说明】
《基因行业蓝皮书》由基因慧联合合作伙伴向公众免费、公开发布电子版及纸质版,均未对外销售或授权任何第三方销售。
《基因行业蓝皮书2024》
产业创新案例(四):鹍远生物


作序专家
黄荷凤院士
卢光琇教授

作序专家
詹启敏院士

作序专家
陈润生院士

作序专家
欧阳颀院士
元英进院士
杨焕明院士
樊春海院士

作序专家
樊嘉院士等
作序专家
樊嘉院士等

作序专家
宁光院士、国家信息中心新兴产业处副处长张振翼等

作序专家
陈润生院士等

作序专家
张泽民教授

作序专家
国家卫健委医药卫生科技发展研究中心原主任李青等

作序专家
国家卫健委医药卫生科技发展研究中心原主任李青、WHO遗传病社区控制合作中心黄尚志教授
拓展阅读
【声明】为了推动生命科技普惠和生物产业发展,基因慧秉持专业、赋能、中立的立场收集、分析及发布相关行业信息;但由于时效性及技术迭代特殊性,所刊登内容仅供研究参考,不作为临床诊疗及投融资等决策依据。本文相关信息不代表基因慧的观点。基因慧平台刊登的原创内容的知识产权为“基因慧”商标拥有者及相关权利人所有;欢迎转载,转载请申请并注明来源。欢迎在基因慧平台合作推广先进的技术、产品及市场成果以及产业规划、行业咨询及市场调研。
基因慧
通过专业咨询平台,提供独立市场洞察解决方案,助力卓越市场表现和生物产业融合普惠。
773篇原创内容