

靳悦 基因慧2023年08月03日 17:51广东

随着我国乃至全球人口老龄化、癌症发病率不断上升、生命科技进步及新疗法的推出,肿瘤精准医疗应用增长势头强劲。本文特梳理基因科技在肿瘤精准医疗方面的应用,从肿瘤早筛、伴随诊断及MRD来分析基因科技应对老龄化及肿瘤防控的技术、解决方案及产业化现状;此外将简单讨论基因科技在生物药研发方面的价值。
一、我国老龄化挑战加剧,肿瘤精准医疗需求增加
截至2021年底,我国 65 岁及以上人口占比突破 14%,在继法国、瑞典、俄罗斯、日本、韩国、美国之后,迈入老龄化国家之列。世界卫生组织(WHO)预估到2050年,全球60岁以上人口占比将达到22%,这个数字是2015年的近2倍(12%),且80%的老龄人口生活在低收入和中等收入国家。老龄化带来公共卫生健康支出挑战,2022年我国卫生健康支出22542亿元,同比增长 17.8%,增长点比教育、科技等高出10余个百分点。
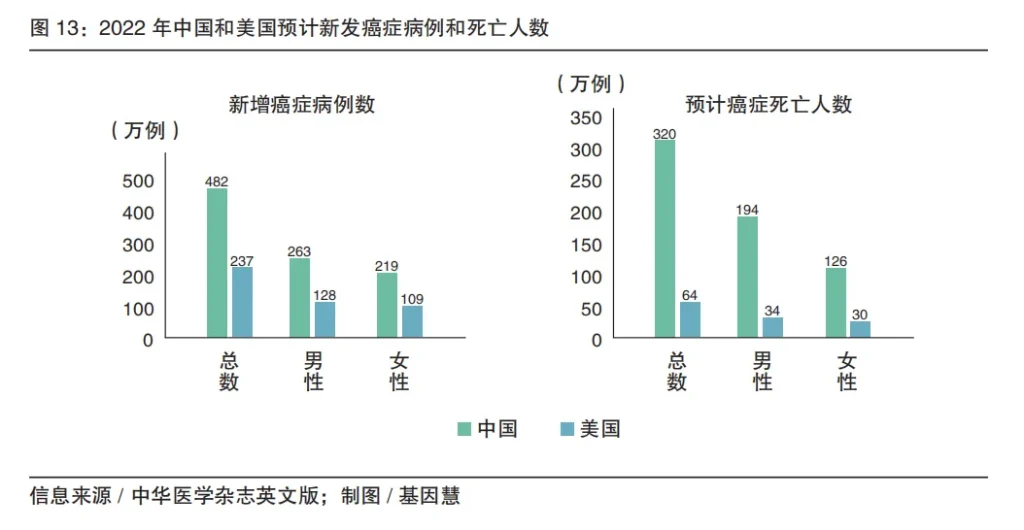
此外,随着急性传染病得到有效的控制和人类平均寿命的延长,癌症已成为严重危害人类健康的重大疾病之一,老龄化加速肿瘤尤其是癌症(近似于恶性肿瘤)的高发及治疗压力。根据《中华医学杂志英文版》预测结果,2022年中国新发癌症病例(482万)大约是美国的两倍,癌症死亡病例(321万)是美国的5倍。我国最常见的恶性肿瘤是肺癌,肝癌、胃癌和食道癌症的负担减少,但肺癌、结直肠癌、乳腺癌和前列腺癌的负担增加。
我国恶性患者的平均五年生存率是西方发达国家的近50%,同时随着我国医疗质量和诊疗能力的提升,特别是近年“健康中国”的战略实施,预防端口迁移,五年生存率从十年前的30.9% 提升到目前的40.5%。但由于大部分肿瘤在早期没有明显的临床表征异常,传统的筛查及检测手段无法及早发现,基因检测以高通量、分子分型、数字化等优势正成为肿瘤早筛、用药指导、预后监测等新兴手段。
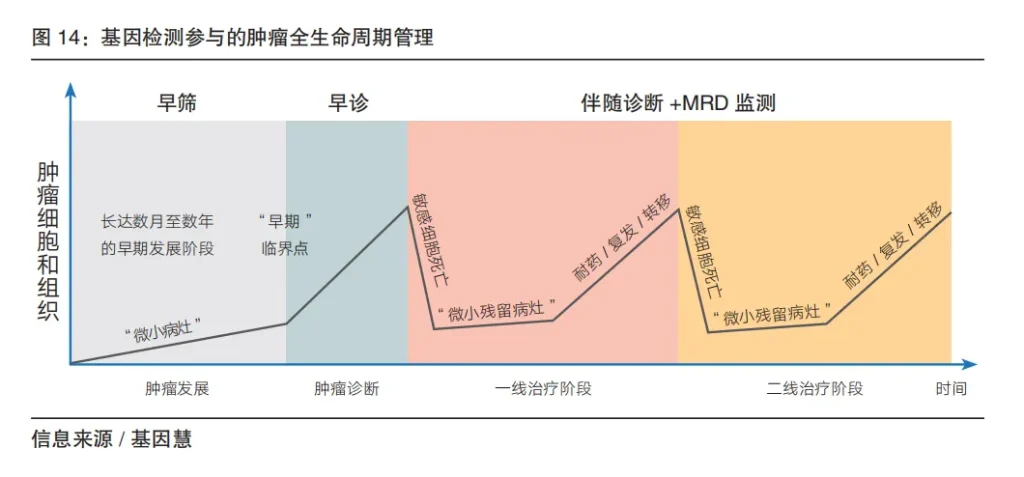
二、肿瘤早筛
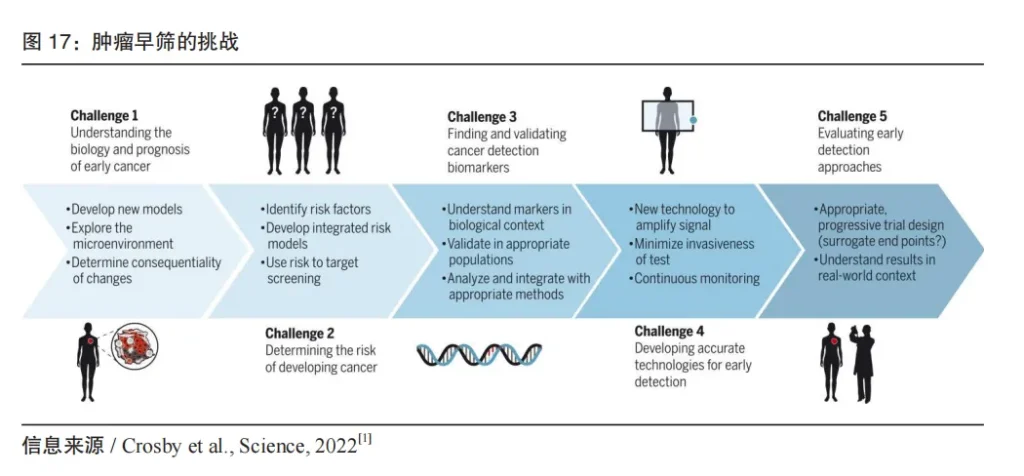
肿瘤(基因)早筛是2022年国内基因检测领域最热门的赛道之一,也是经历高预期回归理性的赛道之一(截止成稿日)。基因慧认为,2023年是肿瘤(基因)早筛赛道产业化的阶段性分水岭,接下来伴随着优秀机构上市、头部机构整合和腰部及以下企业的优化。
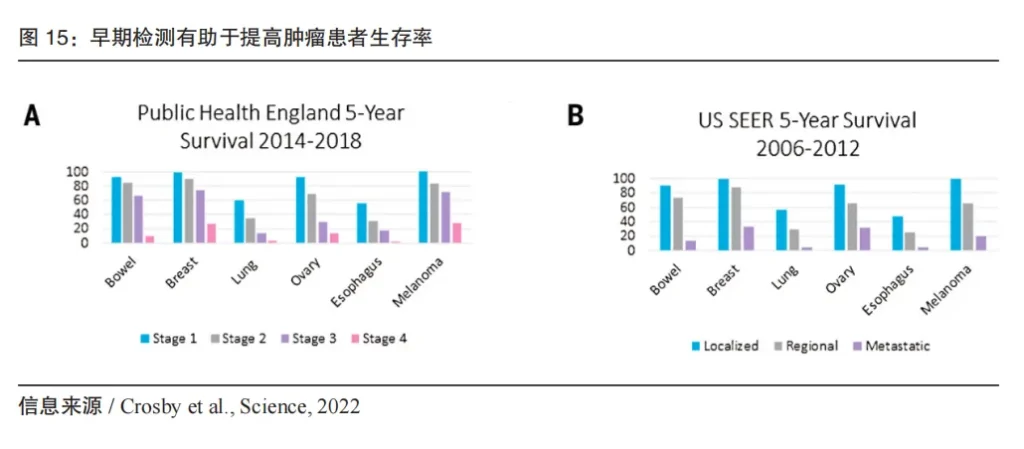
肿瘤早筛的核心需求和价值,是医保基金压力和健康预防的意识提升,通过肿瘤早筛减少癌症发生、提高癌症患者五年生存率。例如,美国推荐50岁以上(后来降至45岁)的人群进行肠镜筛查(到2023年适龄人群肠镜筛查率达到72%),间接导致过去十余年美国结直肠癌的发病率在2000~2010年以每年3%的速度下降(注:由于55岁以上人群比例增长,此数据2010~2019年降至1%);以卵巢癌为例,早发现可将患者的整体生存率从3.5%提升到90%。2014年10月,Cologuard 在获批两个月后,被美国医疗保险和医疗补助服务中心(CMS)纳入联邦医疗保险;2021年10月,北京市医疗保障局签发的《关于规范、调整物理治疗类等医疗服务价格项目的通知》,将基因甲基化检测作为癌症早诊医疗服务项目纳入甲类医保报销范围。
在市场需求层面,从5~10年的长期看,肿瘤早筛的价值不变且将持续提升,特别是周期性的传感染疾病疫情刺激社会及个人的生命健康投资和消费,广泛的受众带来相对高的市场价值和预期估值;同时,未来3~5年,全球性的经济衰退带来一级市场投资趋缓和终端非刚性需求消费的萎缩。
在技术层面,基于基因检测的肿瘤早筛,核心优势是早期发现、可覆盖多癌种、用户依从性好等以及相对高的特异性。主要技术手段是液体活检。液体活检是相对组织活检(例如病理切片等)而言,检测对象包括循环肿瘤细胞(CTC)、循环肿瘤 DNA(ctDNA)、外泌体(Exosome)、细胞外 RNA(exRNA)等。相比其他液体活检方法,ctDNA 以取样相对容易,检测手段相对成熟,并且在检测肿瘤驱动基因突变以及甲基化方面具有高信号丰度和高信号强度等优势,被越来越多用于肿瘤早筛。
根据国家药品监督管理局医疗器械技术审评中心(CMDE)2021年发布的《结直肠癌筛查产品国内外现状及临床评价要求》,疾病筛查类产品的临床试验设计应针对预期的筛查人群进行前瞻性入组,要评价考核检测试剂的临床灵敏度、特异度、PPV(阳性预测值) 和NPV(阴性预测值) 等指标,特别是灵敏度和NPV必须达到较高的水平方能作为筛查产品上市。这对于早筛产品研发及上市提出较大挑战,一是早期肿瘤患者ctDNA丰度(约0.008%)较低,需要提高目前检测技术的极限(约0.1%);二是大样本前瞻性队列所需的资金压力。
因此,从技术上,多种标志物(未来进一步扩展为多组学)联合检测是解决路径之一,例如包括点突变、甲基化、片段化特征、拷贝数变异、蛋白检测等维度,其中最为典型的是“基因点突变+甲基化+蛋白检测”。以2014年FDA批准的全球第一个结直肠癌基因筛查(联合粪便隐血检测)的产品Cologuard为例,采用“NDRG4和BMP3基因的甲基化KRAS 基因的点突变+大便隐血中的血红蛋白检测”,实现结直肠癌筛查92.3%的灵敏度;2020年我国首个获批的肿瘤早筛产品“常卫清”采用类似的技术路径,即基于“体外粪便样本中的KRAS 基因突变+BMP3和NDRG4基因甲基化+血红蛋白检测”,实现4245例入组前瞻性临床试验的结直肠癌高风险人群筛查灵敏度95.5%,阴性预测值99.6%。
除了多靶点多维度检测之外,超深度靶向测序ctDNA突变仍是解决低丰度富集的基础路径(同时带来不可减少的成本);对于肿瘤组织溯源的难题,通过组织特异性的甲基化图谱和循环蛋白标志物等手段来辅助定位。
从应用层面,根据美国临床肿瘤学会(ASCO)官网推荐的重点需早筛的癌种包括:乳腺癌、结直肠癌、宫颈癌、子宫内膜癌、肺癌、前列腺癌等(注:更多内容可参考《2021 肿瘤基因及分子检测蓝皮书》),主要针对较高遗传风险和环境风险的人群进行患病评估和高危人群的鉴别。
肿瘤早筛的应用场景覆盖医疗机构院内检测,第三方医学检验所收样检测,院外体检中心等以及直接面向消费者的居家便捷检测。根据上市机构的商业模式和业绩表现,肿瘤早筛产品现阶段尚处于商业化早期,仍以面向院内患者和院外的高危人群为主,定价相对偏高,特定癌种早筛单价约为2000~4000元,泛癌种早筛约为10000元。国内大部分需患者自费支付,因此目前以诺辉健康为代表的机构在体检等To C渠道获客占有优势。未来,随着社会整体的健康管理水平的提升及医疗保险体系的完善,国家医保和商业保险公司有望为更多有需求的人群提供支付支撑,这有助于进一步扩大市场空间。
国内肿瘤早筛的热度在2020年快速高涨,和瑞基因(代表产品莱思宁)和泛生子(产品 HCCscreen )在肝癌早筛做了早期探索,2022年和瑞基因还推出了多癌种早筛产品(全思宁)。2021年,诺辉健康的常卫清获批给市场注入极大动力,类似获批产品包括锐翌生物的常易舒(检测SFRP2和SDC2基因甲基化)、艾米森的艾长康(检测SDC2和TFPI2基因甲基化)以及早期获批的康立明生物的长安心(检测SDC2基因甲基化)等。
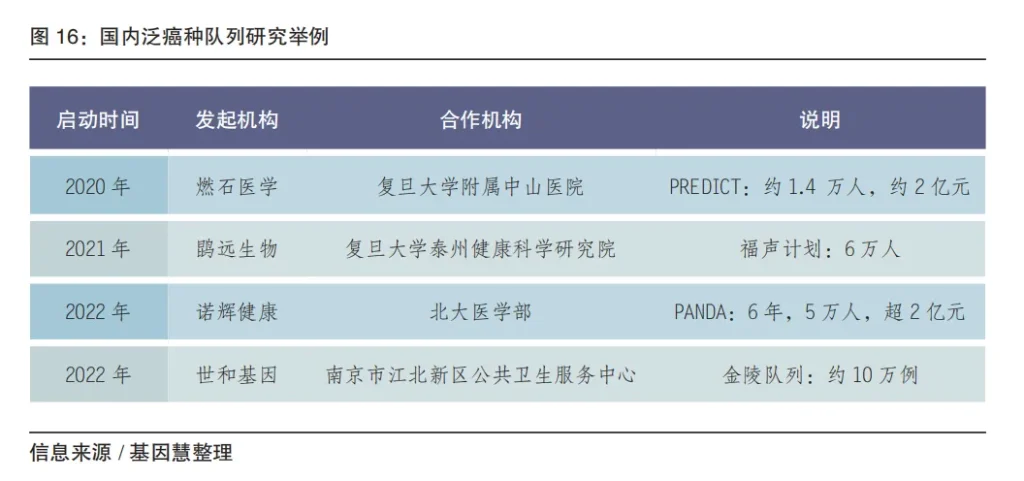
华大基因布局肿瘤早筛领域,以华甘宁、华常康、华消全分别对应肝癌、结直肠癌和泛癌种。燃石医学、鹍远生物、世和基因、和瑞基因等优秀机构纷纷布局万人计、亿元级的前瞻性队列研究。目前存量市场增长乏力(尤其是“院内”),除了拓展体检中心、线上销售等渠道外,也从单癌种逐步扩展到泛癌种。
肿瘤早筛与伴随诊断的模式不同,国内外企业均在早期商业化阶段。2020-2021年对标产品 Cologuard和广阔的受众市场逻辑带来较高估值,2022年因为市场不及预期带来资本的理性回归,2023年继而由于高额的销售费用、大规模队列项目成本压力带来又一轮产业优化周期。
Exact Science的2022年销售费用高达8.46亿美元,但收入/销售费用比2.46,这个数字对于国内上市企业是非常大的挑战。短期控制销售费用的效果,远不及提高收入/销售费用占比,即突破收入增长瓶颈。针对这点,以诺辉健康为例,据公开资料,一方面选择与阿斯利康合作,共同在中国大陆地区公立医院、药店和互联网医院推广常卫清,通过与京东健康、平安健康医疗、云鹊医及中国邮政等合作,提升早筛产品在临床、消费者及保险市场的渗透率;另一方面与Prenetics合作将常卫清销往港澳台以及“出海”东南亚国家或地区,在中国合作渠道包括快验保、港怡医院、香港体检及尚至医疗等超过50家机构;2023年,鹍远生物与马来西亚医疗器械公司Vanguard MedTech达成战略合作,从结直肠癌出发共同拓展马来西亚的肿瘤早检市场,推动鹍远生物的肠癌多基因甲基化检测技术ColonAiQ®落地马来西亚;同年,鹍远生物联合美国突破基因组学(Breakthrough Genomics)公司在加利福尼亚州启动癌症早期筛查和检测服务,运用鹍远生物自主研发的胰腺癌液体活检技术 PDACatchTM 服务广大患者。
在大规模前瞻性队列的研究充分性和资金必要性的难点上,一方面可以参考鹍远生物与扬州市邗江区,世和基因与南京江北新区合作的案例,政企合力聚集高端技术、高端人才和生产制造基地,成为生物健康产业聚集地的同时造就当地民生工程;另一方面,通过引进国有资本、跨界头部机构战略资本是解决各大企业当下“输血”大关的重点。除此之外,技术上的不足,随着检测技术、算法和大规模人群队列的成果转化,长期上不构成阻力;技术迭代带来临床应用场景的可及性以及进而市场准入的加速,其中的关键是延长生存时间,开拓市场同时等待市场逐步成熟。
截止成稿日,鹍远生物自主研发的 PDACatchTM 检测获得FDA的突破性医疗器械认定。PDACatchTM是一种新型的基于DNA甲基化、用于检测高风险人群中胰腺癌和癌前病变的液体活检检测技术。这体现出我国在癌症早筛领域的技术先进性以及在胰腺癌等未来更多癌种甚至泛癌种早筛市场的广阔前景。
三、肿瘤伴随诊断
伴随诊断(Companion Diagnostics, CDx)从英文直译而来,即伴随着治疗而进行的诊断;从作用上可近似理解为用药指导,在基因层面(伴随诊断不限于基因)隶属于药物基因组。
根据 FDA 的定义,伴随诊断用于确定哪些患者最可能受益于特定的治疗产品、可能因治疗而发生严重的副作用以及监测治疗反应。归纳而言,是将治病理念从传统形态学分型(按发病位置)转变到分子分型(按发病机制),实现我国传统中医的“异病同治”或“同病异治”。
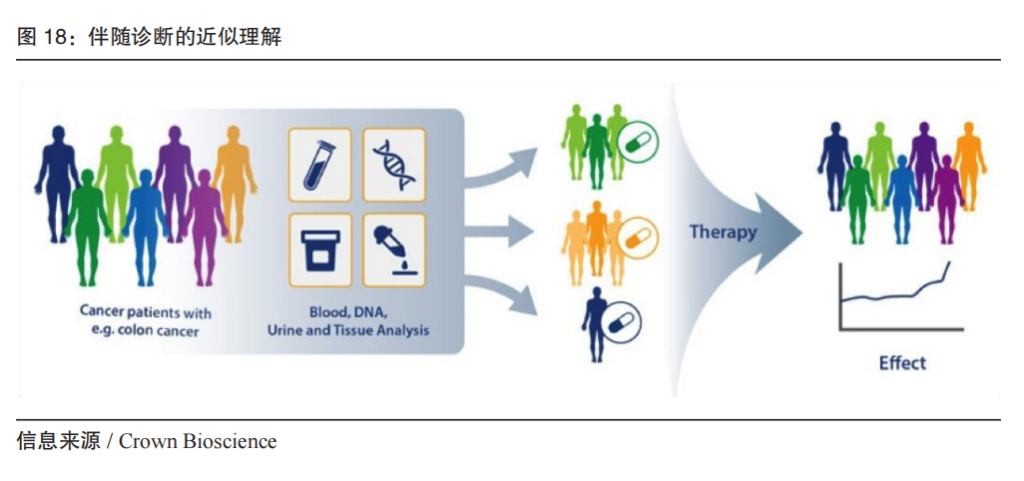
伴随诊断的技术基础是分子分型。组织分型和分子分型是肿瘤伴随诊断期间进行分型的主要方法,组织分型主要依据病理切片手段,而分子分型主要依据 NGS 测序手段。肿瘤分子分型通过对肿瘤驱动基因变异的检测和特征谱的解析,而单细胞测序平台高通量,高分辨率的优势能够更加有效地解析细胞间的异质性,精确地诊断肿瘤亚型并指导进行精准治疗,挖掘精准并且特异地关键评价指标。
根据FDA官网统计,截止2023年5月,FDA 总计批准(Cleared or Approved)针对61种药物(和药物组合)的53种伴随诊断试剂上市,主要集中在抗肿瘤领域,特别是非小细胞肺癌、乳腺癌、结直肠癌等。在技术平台方面,2011年以前以免疫组化(IHC)和原位杂交(ISH)为主;2011年至今,PCR成为FDA批准的CDx 中占比最大的技术(近40%);2016 年以后,NGS 技术的数量和比例逐步增加。
产业机构方面,罗氏诊断通过收购 Ventana 和 Foundation Medicine 成为CDx布局最广的头部机构,其次是安捷伦(旗下Dako)和凯杰。基于NGS技术的CDx 将是未来竞争最激烈的领域,目前主要厂商包括罗氏诊断、赛默飞和因美纳。根据2022年公开财报,罗氏诊断的营收约为200亿美元(177 亿瑞士法郎),同比增长3%;伴随诊断和高端染色带来病理诊断业务11%的增长率。与此同时,2022年罗氏诊断优化了FMI 中国团队。我国NMPA批准的海外企业伴随诊断试剂为个位数,NGS伴随诊断试剂均来自国产厂商,因此 CDx 在我国的发展单独阐述。
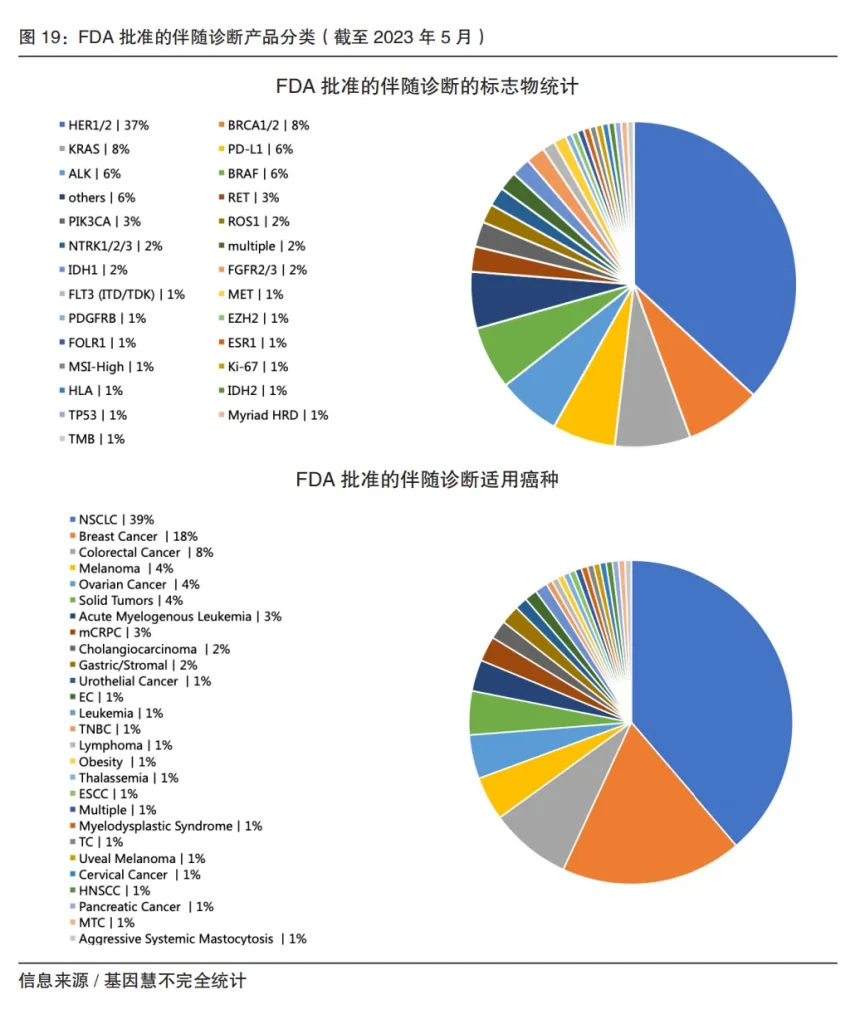
我国伴随诊断起步于2018年,近几年发展迅速,主要是在 NGS 技术平台方面。根据《新型抗肿瘤药物临床应用指导原则(2022 年版)》,57 种靶向药需要伴随诊断,占据近50%。
- 2018年1月,我国首个伴随诊断产品获批:艾德生物 Super-ARMS® EGFR(基于PCR);
- 2018年7月,我国首个NGS肿瘤伴随诊断产品获批:燃石医学的人EGFR/ALK/ BRAF/KRAS 基因突变联合检测试剂盒(可逆末端终止测序法);
- 2018 年 11 月,我国首个 NGS 泛肿瘤伴随诊断获批:艾德生物的人类 10 基因突变联合检测试剂盒;
- 2019年 6 月,北京市将伴随诊断项目纳入其乙类医保;
- 2019年 8 月,我国首个PD-1单抗伴随诊断获批:罗氏诊断的PD-L1 IHC 22C3 pharmDx;
- 2020年 8月,国家药品监督管理局医疗器械技术审评中心(CMDE)发布首个伴随诊断的指导原则《已上市抗肿瘤药物的伴随诊断试剂临床试验指导原则(征求意见稿)》;
- 2021 年 11 月,国家药监局发布《抗肿瘤药物的非原研伴随诊断试剂临床试验注册审查指导原则》、《使用体外诊断试剂境外临床试验数据的注册审查指导原则》;
- 2022 年 3 月,首个国产 PD-L1 检测试剂盒(免疫治疗药物伴随诊断)获批;
- 2022 年 6 月,CMDE 发布《与抗肿瘤药物同步研发的原研伴随诊断试剂临床试验注册审查指导原则》。
根据基因慧不完全统计,NMPA 批准的伴随诊断产品超过40%集中在肺癌(尤其是非小细胞肺癌),近30%是肠癌,14%是乳腺癌,小癌种对应产品极少。非小细胞肺癌伴随诊断标志物集中在T790M点突变、ALK/ROS1 基因融合等热点变异。产业端侧重产品准入逻辑,与临床需求有一定的差距,需要进一步研发差异化伴随诊断及靶向药。
目前CDx的获批IVD产品数量有限,且适应症集中,因此市场上大部分仍以 LDT(实验室自主研发的检测项目)形式在院外开展,当下亟需 LDT 的规范性监管和指导细则出台。长远来看,肿瘤伴随诊断产品伴随分子标志物发现而发展,市场动力之一是靶向药研发合作。FDA批准的 cobas EGFR Mutation Test v2、FoundationOne CDx、FoundationOne Liquid CDx、ONCO/Reveal Dx Lung & Colon Cancer Assay 等伴随诊断试剂可以针对多种(3-5 种)治疗药物且不断扩充范围,为后续多癌种、泛癌种伴随诊断及治疗提供了参考。
四、MRD检测
MRD 指的是微小残留病灶(Minimal Residual Disease, MRD)或分子残留病灶(Molecular Residual Disease)。早在2007年,英国贝尔法斯特女王大学的Alexandra Irvine和Mary Mcmullin对此做过描述,在慢性髓系白血病患者接受治疗后,一小部分会复发,复发源于持续存在的、含量极低、仅能通过分子检测的恶性肿瘤细胞,即MRD。
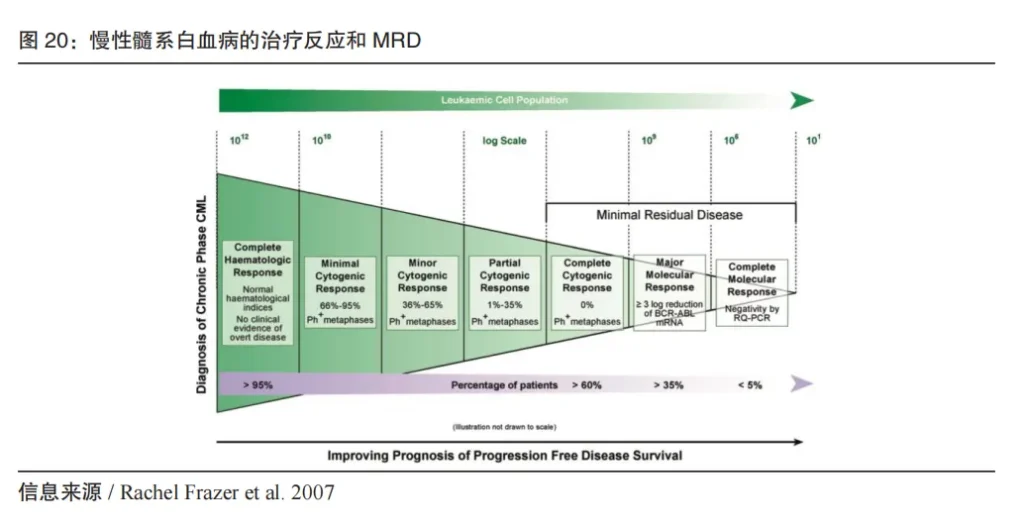
MRD检测有时也归于广义的伴随诊断,但因为作为围术期治疗效果和复发监测的重要方法,对早中期的癌症患者预后监测非常重要,因此也单独成为一类产品。MRD比传统影像学发现疾病复发平均提前5.2个月。四川大学华西医院胸外科的刘伦旭教授在Clinical Cancer Research上发表的非小细胞肺癌前瞻性多中心队列研究(330例I-III期患者,950份血浆样本)成果显示,术后3天和/或1个月ctDNA 阳性(即MRD阳性)可强有力地预测疾病复发风险(HR=11.1),效果优于包括TNM分期在内的临床病理变量。
应用上,MRD 始于血液瘤复发检测,逐步往实体瘤应用。
- 2016年,国际骨髓瘤工作组发布多发性骨髓瘤中 MRD 的评估共识;同年,NCCN 在多发性骨髓瘤的临床指南中增加了MRD 检测;
- 2017年,斯坦福大学 Maximilian Diehn 教授在Cancer Discovery上发表基于ctDNA 的 MRD检测技术;
- 2018年,FDA 批准了首个基于 NGS的MRD产品(Adaptive Biotechnologies 的 clonoSEQ)用于急性淋巴细胞白血病或多发性骨髓瘤患者的MRD检测,并于2019年纳入医保;
- 2020年,FDA正式发布MRD用于血液瘤药物和生物制品开发的指南;
- 2021年,NCCN结肠癌指南中认可基于ctDNA的MRD检测可有效地评估复发风险;
- 2023年结直肠癌 CSCO 指南肯定了MRD在II期结直肠癌化疗决策参考价值。
MRD检测头部机构Natera的Signatera 产品采用“WES+定制Panel”的Tumor informed 策略,在2021年3月获得FDA的突破性设备认定,截止2023年3月,在4个应用方向获得美国医保覆盖:II、III期肠癌全程监测、泛癌免疫治疗疗效监测、肌肉浸润性膀胱癌监测和乳腺癌监测。Guardant Health 的MRD产品Guardant Reveal 采用“甲基化+基因突变的固定 Panel”的Tumor agnostic策略,于2021年4月获得纽约州卫生部临床实验室评估项目(CLEP)的许可,用于检测和监测早期结直肠癌患者 MRD。两种方法各有优劣。
2021年,吴一龙教授等专家达成了我国首个《肺癌MRD的检测和临床应用共识》,提出早期非小细胞肺癌患者根治性切除术后,MRD阳性提示复发风险高,建议每3~6个月进行一次MRD监测;2019年,华大基因与Natera签订协议,独家技术引进Signatera MRD检测。目前尚未有MRD检测产品在我国获批,同时10余家肿瘤基因检测机构发布了近20种 MRD 检测产品。
由于MRD聚焦早中期癌症病人预后复发监测等,与当前药物研发策略吻合,目前各大检测机构与药企合作MRD来加速新药研发,关键点包括LDT细则落地和MRD应用的产品准入。
【声明】为了推动基因及数字生命健康科技推广、产业发展及政产学研用连接,基因慧秉持专业、赋能、中立的立场收集、分析及发布相关信息。但由于时效性及行业特殊性,所刊登内容仅供研究参考,未经说明不作为决策依据;本文相关信息不代表基因慧的观点;基因慧平台刊登的原创内容的知识产权为“基因慧”商标拥有者及相关权利人所有;欢迎转载,转载请申请并注明来源。欢迎个人及机构投稿及合作。