

原创 基因慧 基因慧2023年10月07日 20:38福建

2023年9月29日,哈尔滨工业大学王亚东团队联合中国农科院韩天富团队在bioRxiv发表了题为“A telomere-to-telomere genome assembly of Zhonghuang 13, a widely-grown soybean variety from the original center of Glycine max”的研究文章。研究团队首次对中国大豆品种“中黄13”(ZH13)基因组进行了端粒到端粒(T2T)组装,填补了全部393个空白区域,得到了首个无间隙的中国大豆品种T2T基因组(ZH13-T2T)。
一、研究背景
大豆是世界上重要的油料和蛋白质作物之一,为食品和动物饲料提供了超过四分之一的蛋白质。大豆原产于中国,研究起源地区的遗传资源对推动全球大豆育种工作的发展具有重大意义。
“中黄13”是中国育种家于2001年精心选育的大豆品种,适应性广且具有优良的农艺性状,是中国21世纪前二十年推广面积最大的品种。与美国“Williams 82”相比,中国“中黄13”具有更高的遗传多样性。
此前,为了识别与有利性状相关的关键基因和遗传变异,科学家已经对“中黄13”进行了全基因组测序工作。由于既往测序技术在片段长度方面的固有局限性,现有大豆参考基因组仍有数百个间隙(gap),且端粒和着丝粒等高度重复的基因组区域难以精确组装和注释,导致大豆基因组研究面临一定的瓶颈。

图:文章在bioRxiv上的预印版(来源/paper)
2023年9月29日,哈尔滨工业大学王亚东团队联合中国农科院韩天富团队在bioRxiv发表了题为“A telomere-to-telomere genome assembly of Zhonghuang 13, a widely-grown soybean variety from the original center of Glycine max”的研究文章。
研究团队首次对中国大豆品种“中黄13”(ZH13)基因组进行了端粒到端粒(T2T)组装,填补了全部393个空白区域,得到了首个无间隙的中国大豆品种T2T基因组(ZH13-T2T)。相对之前的中黄13基因组,研究人员通过分析和注释发现了大量差异序列、结构变异、重复序列及具有高置信度的新编码基因。ZH13-T2T基因组的组装代表了大豆基因组学的重大进步,为未来大豆分子育种生物学研究和实践提供了宝贵资源。
二、“中黄13”基因组T2T拼接组装
研究人员对中黄13样本进行了二、三代混合测序,共产生了四类测序数据,包括PacBio Hifi测序(数据量:96.89 Gbp)、ONT超长测序(数据量:96.63 Gbp),Illumina全基因组测序(数据量:55.40Gbp)和Illumina Hi-C测序(数据量:106.4 Gbp)。
通过这些测序数据,研究人员使用一种综合多种拼接算法的自研拼接工作流进行了初步拼接。并在此基础上,进一步通过自研算法进行测序片段重比对和统计,发现拼接结果中的存在的gap区域和潜在错拼区域,并利用一种自研迭代式局部拼接方法进行局部拼接,实现了无gap的端粒到端粒(T2T)拼接。最终,拼接产生的中黄13 T2T基因组长度为1,015,024,879 bp,N50长度为52,033,905 bp。
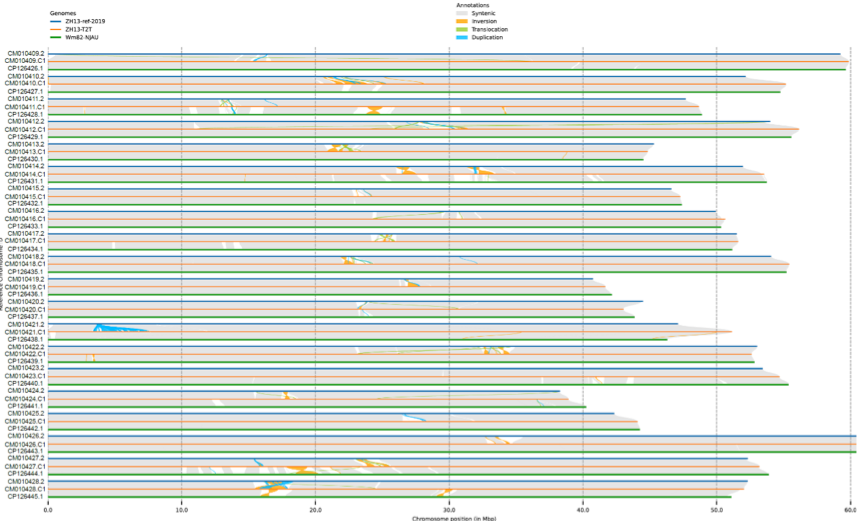
图1:中黄13 T2T基因组与现有中黄13参考基因组和Wm82 T2T基因组的比较
(来源/paper)
研究人员对产生的中黄13 T2T基因组进行了多方面质量评估。其中,前序版本的中黄13参考基因组中存在的393个gap被全部成功填补;20个染色体中的全部40个端粒区域均成功拼接完成,端粒中位数长度达到8449 bp;新版中黄13 T2T基因组的BUSCO指标达到99.8%,碱基质量评分(Merqury质量评分)达到46.441,同时PacBio Hifi和ONT超长测序数据在全基因组上的覆盖度均匀且符合期望值,未发现覆盖度异常区域,显示整个基因组具有较高的拼接质量,并可能不存在显著错拼。
研究人员进一步将中黄13 T2T基因组与前序版本的中黄13基因组和南京农业大学新近发表的Williams 82(Wm82)T2T基因组进行了比较(图 1)。相对于前序版本的中黄13基因组,本研究产生的中黄13 T2T基因组与之共有421个长度超过5Kbp的显著序列差异区域(总长16.3 Mbp)及112个结构变异,包括30个倒置变异、15个易位变异、67个序列重复变异。同时,与Wm82 T2T基因组比较,中黄13 T2T基因组发现了162个长度超过5Kbp的显著序列差异区域(总长23.02 Mbp)和30个结构变异(包括16个倒置变异、7个易位变异、7个序列重复变异)。
三、基因组注释和基因预测
大约57.07%的大豆基因组由注释的重复元件组成。在这些元件中,反转录转座子占38.16%(包括0.12%的SINEs、1.58%的LINEs和36.47%的LTR元件),而DNA转座子占6.72%(图2)。此外,研究人员检测到3.64 Mb的微型卫星,11.58 Mb的小卫星,11.44 Mb的卫星,0.41 Mb的5S rDNA和4.16 Mb的48S rDNA序列。这些串联重复序列占大豆基因组的2.63%(26.65 Mb),明显超过了在中黄13参考基因组序列中观察到的1.03%(10.54Mb)。
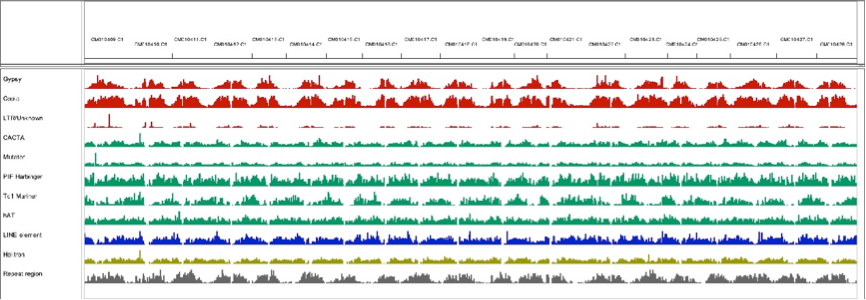
图2:TE 在 ZH13-T2T 基因组中的分布
(来源/paper)
对ZH13-T2T基因组进行从头注释,共获得50,564个高置信度蛋白质编码基因。与ZH13-2019相比,研究人员在gap区域内发现了707个新基因。在此前的gap区域中,研究人员在CM010421. C1染色体的14.84-17.73Mb区域内观察到新发现的基因数量最多,共135个新基因。此外,在这些gap区域内确定了42,668个TEs,300个GmCent-1元素和586个GmCent-2元素。
四、染色体着丝粒鉴定
研究人员使用了串联重复查找工具(TRF)鉴定ZH13-T2T基因组中可能构成着丝粒的重复单体。与之前的研究一致,研究人员发现长度为91和92bp的大量串联重复序列,与着丝粒区域TE的间隙相吻合(图3)。20个着丝粒的平均长度为2.40 Mb,在CM010410.C1染色体上观察到的最长着丝粒(4.42 Mb) 和在CM010421.C1染色体上观察到最短的着丝粒(0.66 Mb)。
值得注意的是,着丝粒长度与染色体大小之间没有显著相关性。此外,着丝粒的相对位置在不同染色体之间存在差异,最小L/S比(长臂长/短臂长)为1.02(CM010415.C1),最大L/S比为2.95(CM010423.C1)。在大豆T2T基因组的着丝粒区共鉴定出8个基因。这些基因主要富集染色质DNA结合、mRNA顺式剪接、组蛋白结合、基底转录因子、剪接体和嘧啶代谢(图3)。
平均而言,着丝粒序列由96.0%的着丝粒卫星DNA(CentC)、着丝粒反转录转座子(CRM)和其他非CRM Gypsy反转录转座子组成。这些成分的比例在不同的着丝粒中差异很大,GmCent-1从0.0%到73.3%,GmCent-2从0.0%到90.4%,CRM从0.0%到2.2%,其他非CRM Gypsy反转座子从7.3%到68.2%不等。几乎所有的着丝粒都富含 CentC。
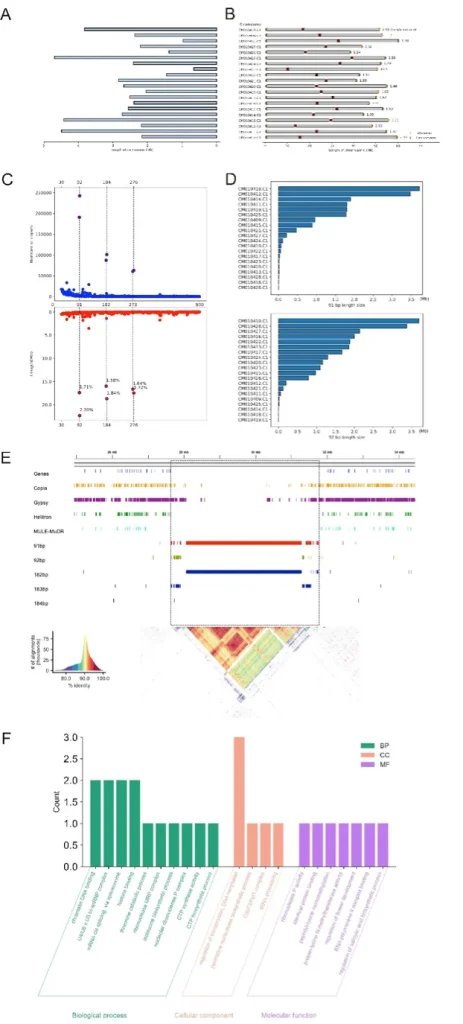
图3:端粒和着丝粒的基因组结构
(来源/paper)
五、研究意义
该项研究是中国大豆品种的首个T2T基因组组装。T2T基因组提供了无间隙的参考基因组,也为全面解码和深入了解大豆基因组中的复杂重复提供了新的机会,这对于植物基因组学研究具有重要的价值。
大豆是一种典型的短日作物,光温反应敏感。花期和生育期是决定大豆生态适应性的最主要因素。以往报道了大量的花期和生育期数量性状基因座和显著关联位点,但在挖掘主效基因时候选区域往往较大,并且已有的参考基因组存在遗漏候选基因的风险。文章在T2T-ZH13参考基因组注释揭示了大量新基因,为高效挖掘目标农艺性状候选区域主效基因提供了新的分子靶点。作为我国推广面积最大的品种之一,ZH13-T2T基因组将为今后大豆分子育种工作提供了宝贵资源。
【声明】为了推动基因及数字生命健康科技推广、产业发展及政产学研用连接,基因慧秉持专业、赋能、中立的立场收集、分析及发布相关信息。但由于时效性及行业特殊性,所刊登内容仅供研究参考,未经说明不作为决策依据;本文相关信息不代表基因慧的观点;基因慧平台刊登的原创内容的知识产权为“基因慧”商标拥有者及相关权利人所有;欢迎转载,转载请申请并注明来源。欢迎个人及机构投稿及合作。